
MS-HRM qPCR を用いた CpG メチル化の定量
Pedro Quintas PhD & Constantine Garagounis PhD
Introduction
DNA メチル化は、核酸塩基 (主にシトシン) がメチル基の付加によって修飾されるエピジェネティック プロセスです。このメカニズムは、遺伝子発現、発達、疾患の調節を含む細胞プロセスにおいて重要な役割を果たします。このアプリケーション ノートでは、メチル化感受性高解像度融解 (MS-HRM) qPCR を使用して特定の遺伝子座の DNA メチル化を分析するワークフローについて説明します。
この方法は、DNAを亜硫酸ナトリウム[1]で処理することで、メチル化されていないシトシンをウラシルに変換し、メチル化されたシトシンはそのまま残します。これらのウラシルは、PCR増幅中にチミンに置き換えられます(図1)。したがって、メチル化されたDNAテンプレートから生成されたアンプリコンには、メチル化されていないDNAテンプレートから生成されたアンプリコンよりも、より安定したC-G塩基対が多く含まれています。その結果、メチル化されたターゲットの融解温度が上昇します。
この融解温度のシフトは、高解像度融解(HRM)qPCR [2]を使用して決定されます。このプロセス中、混合物中に存在する挿入色素がPCR中に生成された二本鎖DNAに結合し、蛍光を生成します。増幅後、反応混合物は融解プロファイルを生成するために上昇する温度にさらされます。温度が生成物の融点に達すると、DNA鎖が解離し、色素が放出され、蛍光の急激な減少が観察され、生成物の融解温度を正確に推定できます。亜硫酸水素塩処理によって生成物の融点がどの程度影響を受けるかは、それに含まれるメチル化シトシンの数の指標となります。
この方法の主な利点は、ハイスループットのワークフローに適応できることであり、したがって、多くのサンプルのメチル化レベルを同時に測定する費用対効果が高く、迅速かつ正確な方法が提供されます。
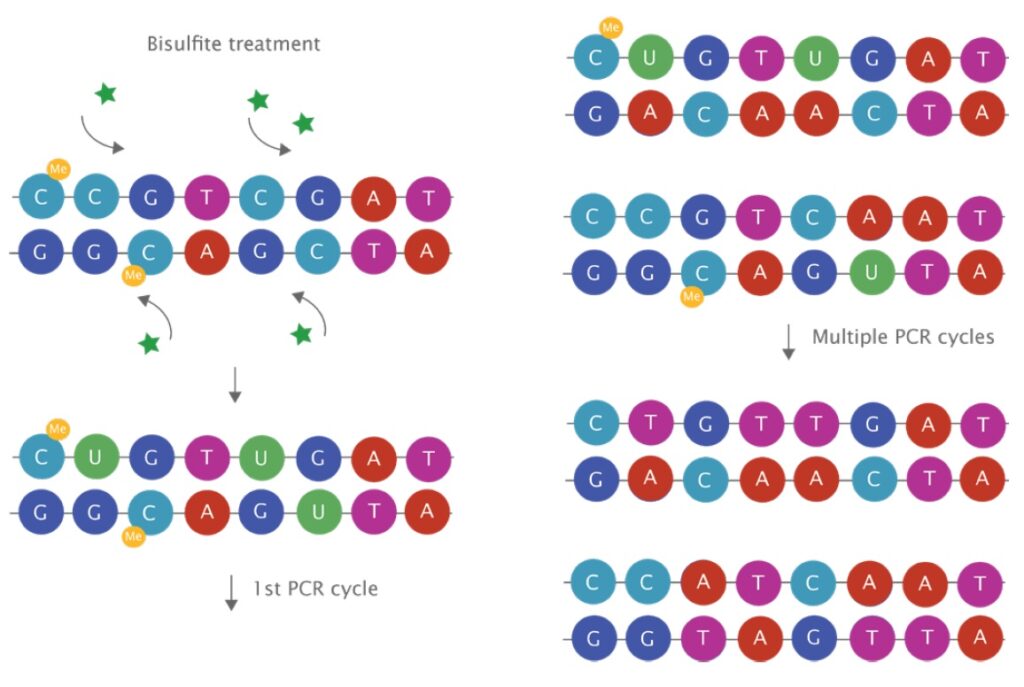
図1:DNAの亜硫酸水素塩処理により、メチル化されていないシトシンがウラシルに変換される
PCR 中、ウラシルは新生 DNA 鎖に組み込むアデニンのテンプレートとして機能します。後続のサイクルでは、これらのアデニンは各鎖のメチル化されていないシトシンに対応する位置にチミンを組み込むためのテンプレートとして機能します。複数の PCR サイクルの後、すべてのメチル化されていないシトシン-グアニン塩基対はチミン-アデニン塩基対に置き換えられます。
Method
PCR Biosystems 社の Clara® HRM Mix と HRM 実験を実行できるサーマルサイクラーを使用して、DNA サンプルのメチル化レベルを測定できます。このアプリケーション ノートでは、ヒト FN3K 遺伝子のメチル化レベルを決定するために使用した実験条件について説明します。
Primer design
プライマーの選択は、MS-HRM (メチル化感受性 HRM) 実験にとって重要です。新しいアッセイを開発する場合、より良い結果が得られるものを選択する前に、いくつかのプライマー セットを試すことをお勧めします。このアプリケーションでは、公開されているプライマー セットを使用しました (表 1 を参照)。新しいアッセイ用のプライマーを設計する場合、いくつかの考慮事項に留意する必要があります。
- アンプリコンは約 100 bp で、プライマー間に 3 ~ 10 個の CpG サイトが含まれている必要があります。メチル化サイトの数が多いほど、メチル化配列と非メチル化配列の Tm 差が大きくなります。ただし、メチル化サイトの数が多すぎると、PCR がアンプリコンの 1 つに偏る可能性があります。
- プライマーの融点は 56~60 °C で、融点の差が 1~2 °C 以内になるように設計します。GC 含有量を 35~65% に保つようにします。
- 各プライマーの 5′ 末端近くに 1 つまたは 2 つの CpG サイトを含めます。
- プライマーは、亜硫酸水素塩処理後、メチル化されたテンプレートと相補的である必要があります。したがって、プライマーには、メチル化されていない C 残基の代わりに T が含まれている必要があります。

表1:この MS-HRM アッセイで使用されるターゲット配列 (亜硫酸水素塩処理後) とプライマー
CpG部位のシトシンはマゼンタでマークされ、亜硫酸水素塩処理により非CpGシトシンから生じたチミンは青緑色でマークされています。プライマーの位置は下線で示されています。[3]から引用。
Template preparation
標準曲線を作成するために、等濃度のメチル化 DNA と非メチル化 DNA (市販) を異なる割合で混合し、必要な範囲をカバーしました。今回は、メチル化の全範囲をカバーしたかったので、0%、20%、40%、60%、80%、100% のメチル化に対応するサンプルを準備しました。次に、Promega MethylEdge® Bisulfite Conversion System を使用して、各サンプル 20 μL を亜硫酸水素塩で処理しました。このシステムは、非メチル化シトシン (C) を脱アミノ化してウラシル (U) を形成しますが、メチル化シトシンには影響しません。同じ処理を、テストするサンプルにも実行しました。
Reaction setup
反応は Qiagen QIAgility ロボットを使用して設定されました。表2に報告されているように、最終容量は20μLです。
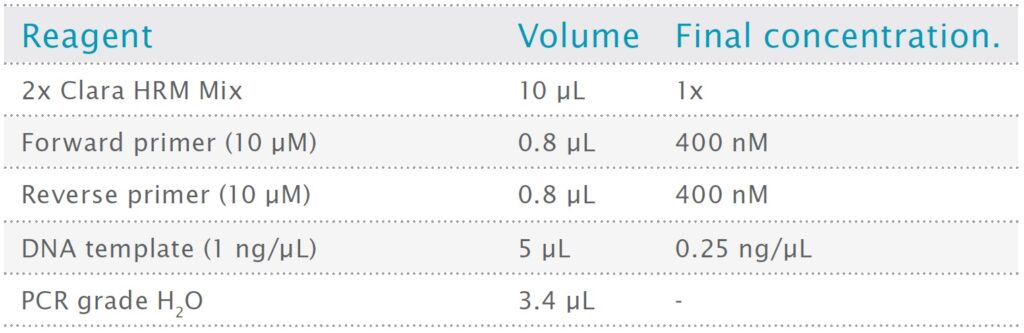
表2:反応のセットアップと構成
Cycling conditions
サーマルサイクリングは、表 3 に概要を示したサイクリング パラメーターを使用して Roche LightCycle 96 qPCR マシンで実行されました。アニーリング温度は MS-HRM 実験にとって重要なパラメーターであり、経験的に決定する必要があります。目標は、各メチル化レベルで融解曲線の良好な分離を得ることです。温度を上げると、プライマーがメチル化テンプレートにさらに特異的に結合します (CpG メチル化部位を含むように設計されていると仮定)。これにより、メチル化レベルが低いテンプレートの分離が向上します。プライマーダイマー形成のリスクが高まるため、アニーリング温度を下げすぎないように注意する必要があります。
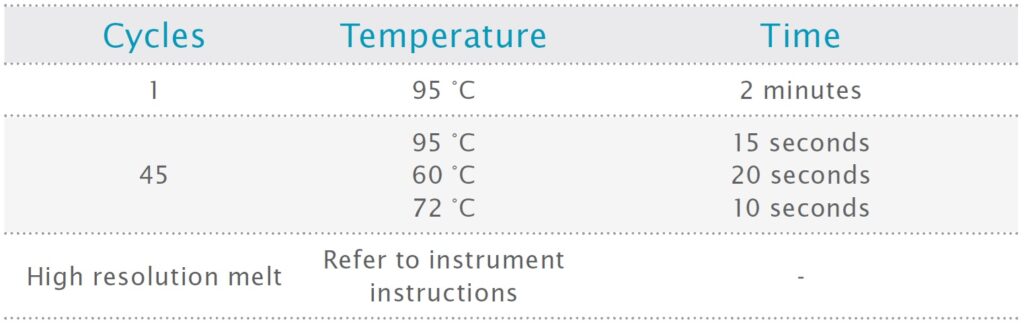
表3:サイクリング設定
Results
結果は、機器ソフトウェアの HRM 機能を使用して分析されました。この機能は、開始蛍光レベルと終了蛍光レベルを正規化し、融解曲線を融解ピークに変換します (図 2)。次に、各ピークの最大蛍光をメチル化レベルに対してプロットして標準曲線 (図 3) を作成し、これを使用してテストサンプルのメチル化レベルを推定できます (表 4)。
PCR 産物は、配列決定によってさらに分析され、メチル化のレベルを確認し、メチル化されたシトシン残基を特定することができます。
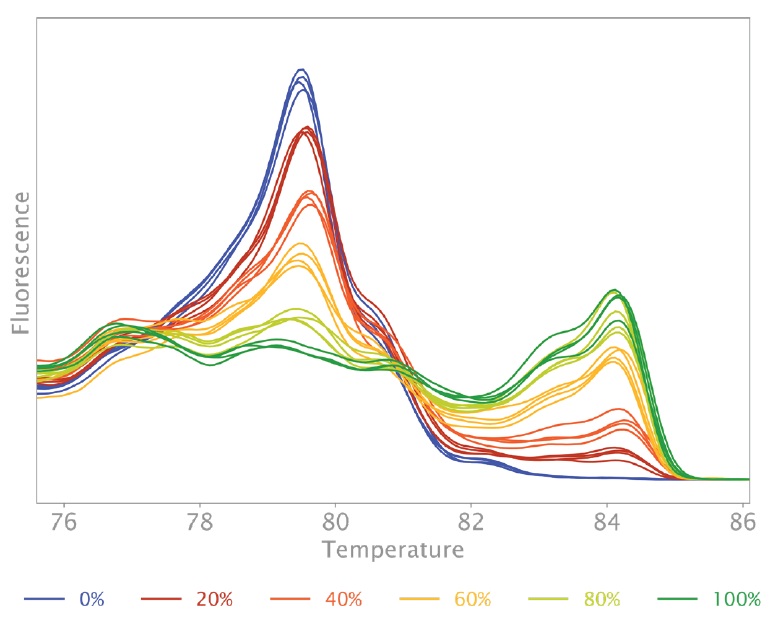
図2:FN3K 遺伝子標準曲線の MS-HRM 解析から得られた融解ピーク
このグラフは、メチル化率が既知のさまざまなサンプルを使用して作成されました (下のグラフに示されています)。
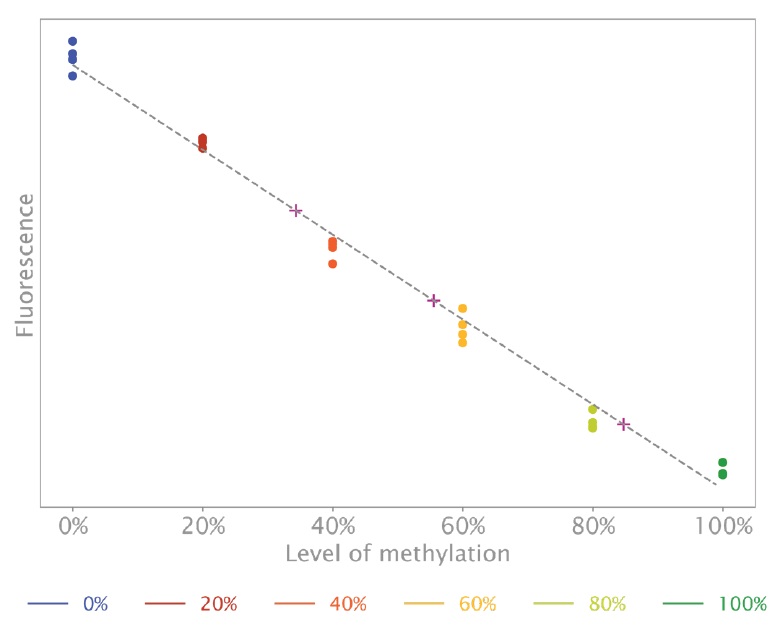
図3:メチル化率の定量化
標準曲線は、79.5 °C での標準曲線サンプルの蛍光を既知のメチル化率 (表示) に対してプロットすることによって取得され、テストサンプルの推定メチル化は + 記号で表されます。
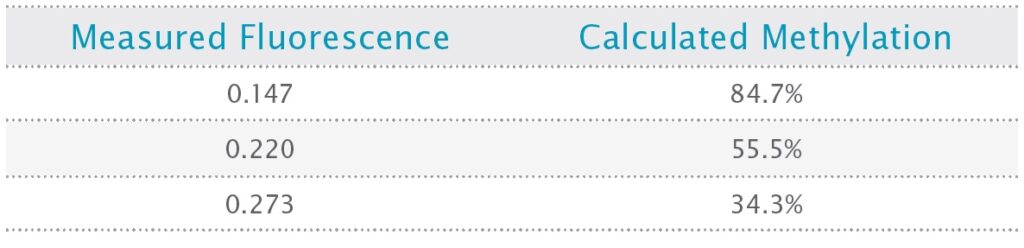
表4:試験サンプル中のメチル化レベルの計算値
Discussion
ゲノム DNA のシトシンメチル化は、発達と疾患に重大な影響を及ぼします。メチル化に敏感な高分解能融解分析は、DNA サンプルのメチル化レベルを迅速、高感度、正確に定量する方法を提供します。PCR Biosystems 社の Clara® HRM Mix は、SNP ジェノタイピングと MS-HRM の両方においてクラス最高のパフォーマンスを提供します。
Product use
Clara® HRM Mix を含む PCR Biosystems 社製品単独では診断結果は得られず、研究用途のみに提供されています。ただし、すべての製品は ISO 13485 準拠の管理システムの下で製造されており、該当する国の法律で許可され、アッセイ自体の臨床検証が済んだ後は、分子診断アッセイのコンポーネントとして使用するのに適しています。
どの製品がお客様のアプリケーションに最適かご相談されたい場合、またはClara® HRM Mix を MS-HRM 分析に使用する方法についてさらに技術的なアドバイスが必要な場合は弊社までお問い合わせください。
References
- M. Frommer, L. E. McDonald, D. S. Millar, C. M. Collis, F. Watt, G. W. Grigg, P. L. Molloy and C. L. Paul, “A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands,” Proc Natl Acad Sci U S A, vol. 89, no. 5, pp. 1827-31, 1992.
- T. K. Wojdacz and A. Dobrovic, “Methylation-sensitive high resolution melting (MS-HRM): a new approach for sensitive and high-throughput assessment of methylation,” Nucleic Acids Res, vol. 35, no. 6, p. e41, 2007.
- D. Hussmann and L. L. Hansen, “Methylation-Sensitive High Resolution Melting (MS-HRM),” in DNA Methylation Protocols. Methods in Molecular Biology, vol. 1708, J. Tost, Ed., New York, NY, Humana Press, 2018.