
qPCRテクニカルガイド
製品ラインナップは下記よりご覧ください
1. イントロダクション
1.1 qPCRとは?- その仕組み
定量的 PCR (qPCR) は、核酸ターゲットの検出および定量化のための最も重要な方法の 1 つです。この手法はリアルタイムqPCRとも呼ばれ、RT-qPCR(逆転写qPCR)とは別物です。従来のPCRと同じ基本的な試薬、酵素、サーマルサイクリング条件を用いますが、反応中の生成物量に比例して蛍光を発生する試薬も追加されます。次に、蛍光光度計を備えた特殊なサーマルサイクラーを使用して、シグナルをリアルタイムで記録します (図1)。この蛍光は、任意蛍光単位(AFU)または相対蛍光単位(RFU)で測定されます。
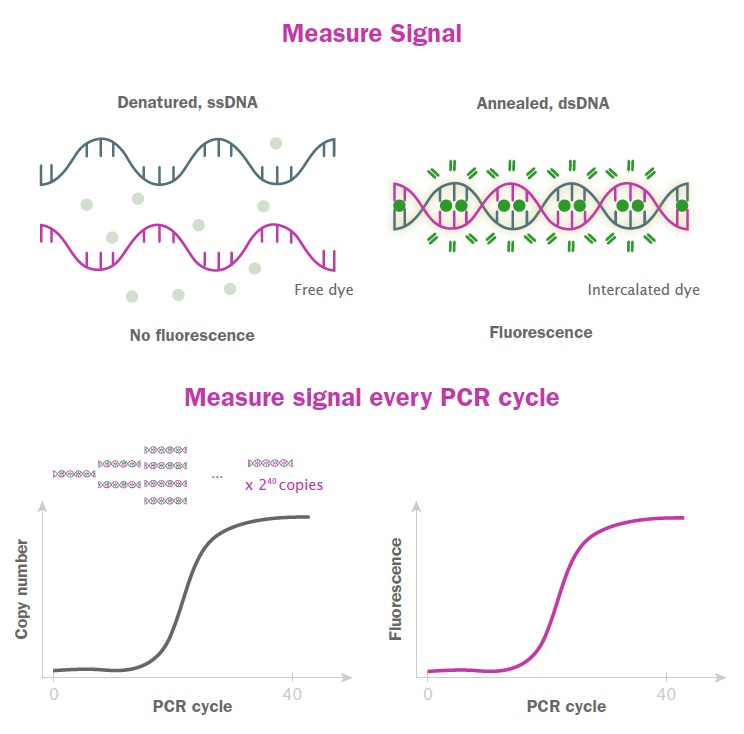
図1:qPCRにおける色素ベースの蛍光シグナル生成
従来のエンドポイント PCR では、最初のターゲット存在量の半定量的な評価しかできません。これは、DNA ポリメラーゼの生成物の阻害と試薬の枯渇により、PCR 効率が後のサイクルで維持されないことが原因です。逆に、qPCR では生成物の蓄積をリアルタイムでモニタリングできるため、より正確な定量的情報を得ることができます。これは、qPCR反応の蛍光強度をPCRサイクル数に対してプロットすることで生成される増幅曲線から抽出されます。
典型的なqPCR増幅曲線はシグモイド曲線です。最初は、蛍光は線形グラウンド、つまり開始期にバックグラウンドノイズ以下で増加します。産物が指数関数的に蓄積するにつれて、蛍光は比例して増加し、バックグラウンドノイズレベルを超えます。PCR試薬が枯渇するまで、蛍光は各サイクルごとに対数線形に増加し続けます。その後、反応が遅くなるか停止するため、サイクリングプログラムの終了まで蛍光蓄積はプラトー期に達します(図2)。増幅曲線のこの形状に異常がある場合は、反応に問題があることを示しています。このような異常の解釈方法と修正に必要な手順の詳細については、セクション9の「トラブルシューティング」を参照してください。
qPCR 増幅曲線から得られる最も価値のある測定基準は、いわゆるしきい値サイクル (Ct) または定量化サイクル (Cq) です。これは、ターゲット産物によって生成された蛍光シグナルがバックグラウンドノイズを上回る任意のしきい値レベルを超えるPCRサイクルに相当します(図2)。この Cq 値は初期ターゲット濃度に比例し、異なるサンプル間でターゲットの量を比較するために使用できます。あるいは、既知のターゲット量を使用したリファレンスサンプル Cq の標準曲線と比較して、初期テンプレートの定量化を可能にすることもできます。
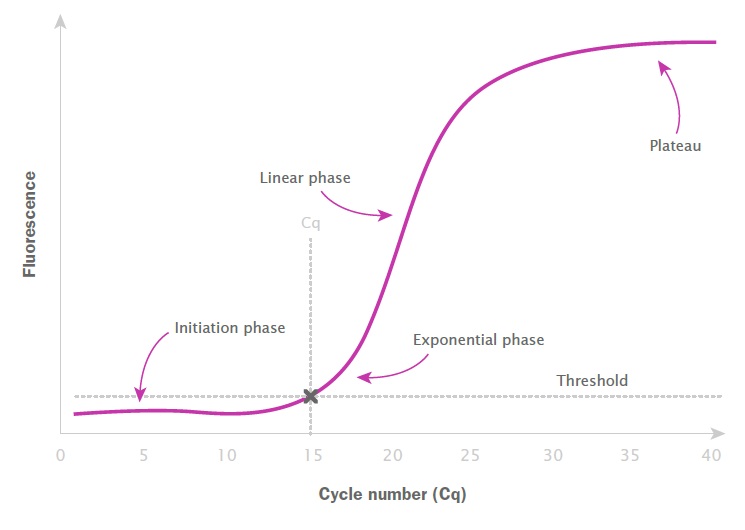
図2:一般的なqPCR増幅プロット
2. qPCRの検出方法
色素ベースの検出とプローブベースの検出という 2 つの異なる化学戦略を使用して、蛍光を介した生成物の蓄積のリアルタイムモニタリングを可能にすることができます。
2.1 色素ベースの検出
PCR産物をリアルタイムで検出するこのアプローチは、二本鎖DNA分子にインターカレーション可能な蛍光色素を添加することで実現します。この色素自体は溶液中で遊離している状態で一定の蛍光を発しますが、DNA塩基対間に同数の色素分子が重なると蛍光の強度が劇的に増加します。したがって、qPCR反応の開始時には、遊離した色素から一定の蛍光が発せられるのに加え、反応開始時に二本鎖DNAにインターカレーションされている少量の色素(本来は少量であるべき)が蛍光を発します。
PCRが進むにつれて、サンプル中の二本鎖DNA(dsDNA)の量が増加します。対応して、フリーの色素に対するインターカレートされた色素の比率が増加し、蛍光強度も比例して増加します (図3)。数サイクル後、PCR 反応の速度が低下して停止するため、蛍光はプラトーに達し、前述した蛍光のシグモイド曲線が生じます。多くの場合、特に旧式のqPCR装置では、qPCRミックスにパッシブ色素(通常はROX)を追加することで、実行中の蛍光シグナルの変動を標準化します。
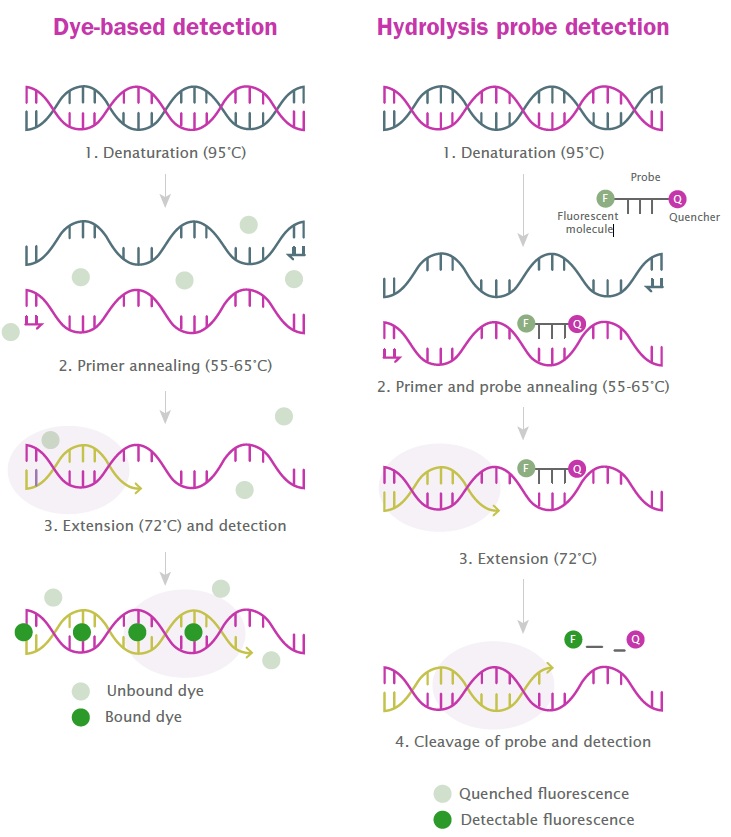
図3:qPCR における色素と加水分解プローブのシグナル生成メカニズム
色素ベースの qPCR のメリットとデメリット
メリット
- この色素は、特別に設計されたプライマーペアと組み合わせることで、任意のターゲット分子を検出することができます。これは、複数の異なる標的をターゲットとするハイスループットの独立した qPCR を、シンプルかつ安価な方法で設計できることを意味します。
- DNA インターカレート色素を使用すると、蛍光シグナルが可逆的に蓄積されます。インターカレーション色素のみが強い蛍光を発するため、DNAの変性は蛍光シグナルの減少につながります。これにより、融解曲線分析を用いて反応の特異性を検証することが可能になります。
デメリット
- 非特異的な蛍光シグナルは、インターカレート色素に結合する任意の dsDNA によって生成されます。したがって、ミスプライミングに起因する偽産物、サンプル中のターゲットと類似した配列の存在、プライマーダイマーの形成、プライマーヘアピン構造の可能性などは、いずれも反応中に偽シグナルの蓄積につながる可能性があります。結果を検証するために、融解曲線分析、エンドポイント産物のゲル電気泳動、またはサンガーシーケンシングを実施する必要があります。
- どの反応でも測定できるターゲットは1つだけなので、マルチプレックス反応は実現できません。サンプル内の複数のターゲットを定量する場合は、対応する数の反応をセットアップする必要があります。
2.2 融解曲線分析
融解曲線解析により、qPCR産物の融点を特定できます。この情報は、qPCR産物の特異性を確立するための指標として有用です。融解曲線解析を行うには、qPCRサイクリングプログラムの最後にサーマルサイクリングステップを追加します。このステップでは、インキュベーション温度を徐々に上昇させ(通常50~55℃から95~99℃)、反応サンプルをゆっくりと変性させます。その間、蛍光は0.1~0.5℃ごとに段階的にカウントされます。温度に対してプロットされた蛍光強度の累積データポイントが融解曲線となります。
特定の生成物を含む反応では、温度がターゲット物質の融点に近づくまで蛍光は一定に保たれ、その後急速にベースラインまで減少します。蛍光が最大値の50%に低下した点が、その分子のTm値とみなされます(図5)。
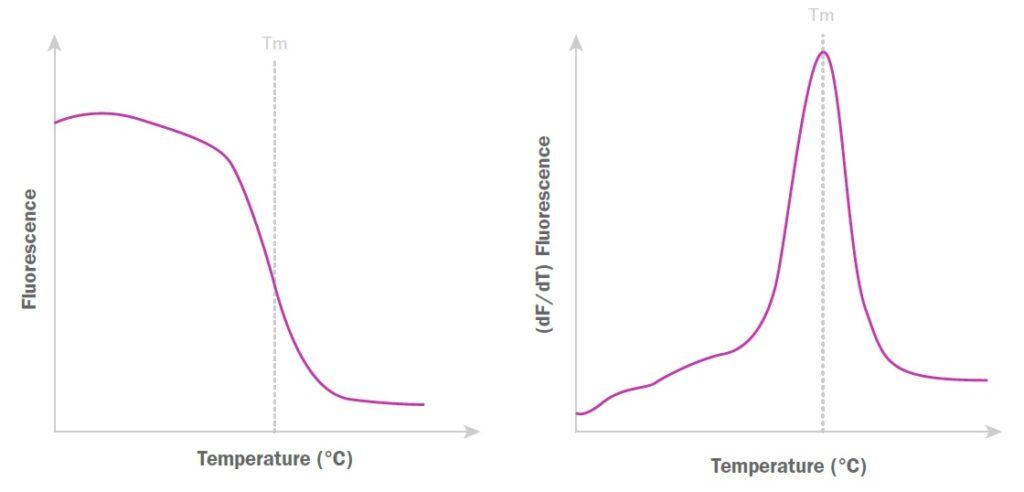
図5:qPCR融解曲線とその結果得られる融解ピークの例
リーディングピーク、歪んだピーク、または複数のピークはすべて、反応中に非特異的な生成物が蓄積していることを強く示しており、テンプレートの定量分析を行う前にさらに調査する必要があります (図 6)。
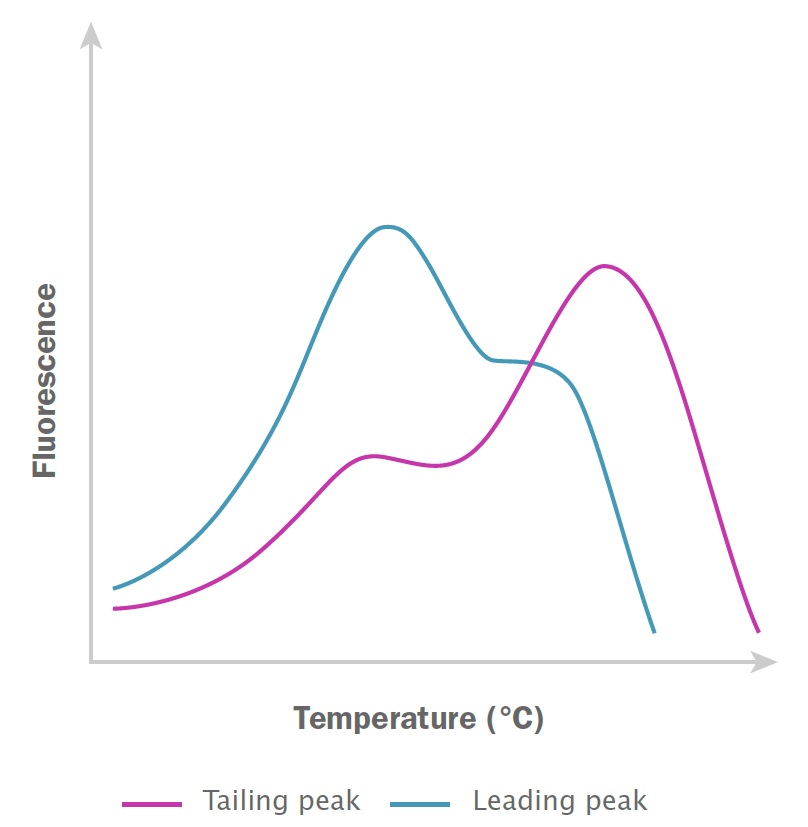
図6:問題のある融解ピーク、テーリングピークおよびリーディングピークの例はプライマーダイマーを示しています
融解曲線分析を行う場合、最初に使用するすべてのプライマーペアを検証することが重要です。最終的な PCR 産物をアガロースゲルで電気泳動し、観察された融解ピークが予想されるターゲット産物のサイズに対応していることを確認します。融解曲線/ピークは特異性の強力な指標ですが、類似した長さと配列を持つターゲットであっても、同じ融解曲線を示す場合があることに注意することが重要です。さらに、異なる試薬、異なるイオン強度、そして異なるサンプルに存在するランダムな夾雑物によって、目的のターゲットが高特異性で増幅されたとしても、観測される融解ピークにシフトが生じる可能性があります(図7)。
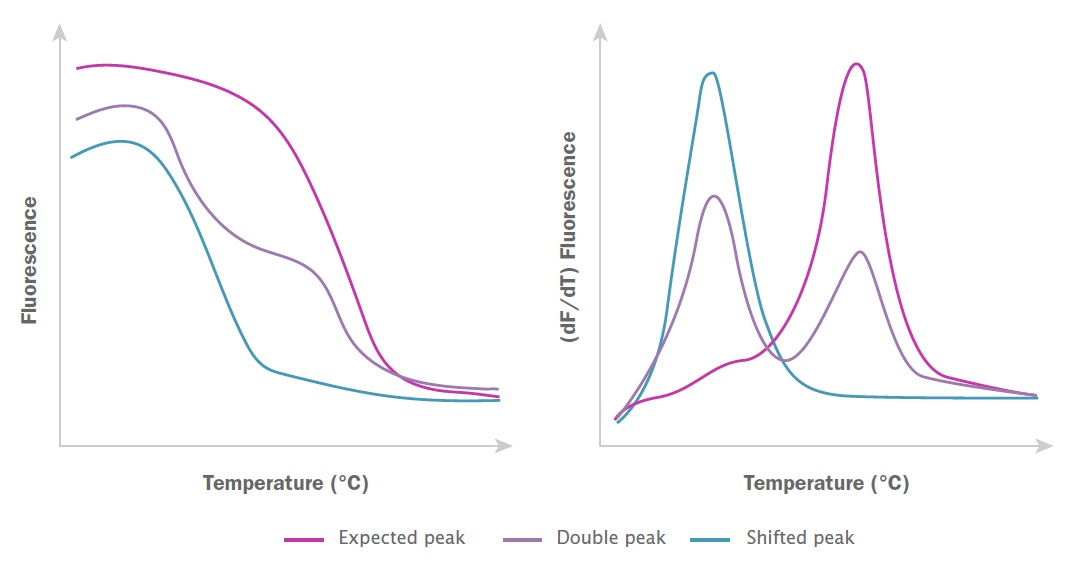
図7:qPCR融解曲線とその結果得られる融解ピークの例
異常な融解ピークは、オフターゲットプライミングやプライマーダイマーまたはプライマーヘアピンの形成によって形成される複数の生成物など、複数の生成物が反応中に蓄積する場合にも観察されます。反応中に複数の産物が存在すると、それぞれ異なる温度を中心とする、通常は異なる融解ピークが複数生成されます(図7)。場合によっては、複数の生成物が二重ピークまたはリーディングピークとして表示されることがあります。プライマーダイマーまたはヘアピンは通常、全長の産物よりも低い温度で現れますが、必ずしもそうとは限りません。
他の産物が存在しない場合でも、特定のアンプリコンが二重ピークを生成する場合があります。これは、アンプリコン配列の異なる領域でGC含量が大きく異なる場合に発生することがあります。このような不均一な塩基対含量により、まずGC含量の低い領域が部分的に変性し、続いてGC含量の高い領域が高温で変性し、最終的に2つの異なるピークが現れます。注目すべき例として、哺乳類のCFTR遺伝子2が挙げられます。この現象はまれですが、qPCR分析を行う際には注意が必要です。
2.3 プローブベースの検出
qPCR プローブには、加水分解プローブ、デュアル ハイブリダイゼーション プローブ、ヘアピン プローブという異なる作用機序に基づいた 3 つタイプがあります。これらのいずれの手法においても、蛍光検出はFRETと呼ばれる物理現象を利用しています。本稿では主に加水分解プローブ(TaqMan®プローブとも呼ばれます)に焦点を当てますが、網羅性を高めるため、他の種類についても簡単に説明します。
Förster Resonance Energy Transfer (FRET)
FRETは、2つの光感受性分子が蛍光に依存しない方法でエネルギーを伝達する物理現象です。この現象において、2つの分子のうちの1つはドナーとして働き、電子励起状態においてもう1つのアクセプター分子にエネルギーを伝達し、これを励起します。アクセプター分子の性質に応じて、このエネルギーは蛍光として放出される場合もあれば、光に依存しない方法でエネルギーを消散させる場合もあります。後者の場合、アクセプター分子はクエンチャーとして作用します。同時に、ドナー分子は蛍光を発することなく非励起電子状態に戻ります。
加水分解プローブ
加水分解プローブを使用したターゲット生成物の検出は、ターゲット分子の一部に相補的になるように設計されたオリゴヌクレオチドプローブを追加することによって実現されます。プローブは共有結合により修飾され、一方の端には蛍光団が含まれ、もう一方の端にはその近傍でのみ蛍光団から放射される光を吸収(消光)する消光分子が含まれます。バックグラウンドシグナルを低減するために、2つ目のクエンチャー分子(プローブ配列の途中に添加)が必要となる場合もあります。また、このプローブは、ターゲット分子の増幅に用いるプライマーペアと同程度のアニーリング温度となるように設計されています。
qPCR反応液に添加した直後は、クエンチャーが蛍光シグナルを吸収するため、蛍光シグナルは生成されません。サーマルサイクリング プログラムが開始されると、プログラムの変性/アニーリング段階でプローブがターゲットとアニーリングします。伸長中、新しい DNA 鎖を生成するポリメラーゼは、5′-3′ エキソヌクレアーゼ活性を通じてアニーリングされたプローブを分解し、反応ミックス中のクエンチャー分子と蛍光分子を遊離します。これにより、これら2つの分子間の距離が大幅に広がるため、蛍光分子は刺激を受けると検出可能な蛍光シグナルを発します(図3)。
各サイクルが繰り返されるにつれて、ターゲット DNA 分子の量が増加すると、分解されるプローブの量が増加し、放出された蛍光分子から放出される蛍光シグナルがますます増加します。これにより、各サイクルから一連のデータポイントが生成され、予想されるシグモイド曲線が得られ、そこからCqサイクル値が算出されます。色素ベースの検出と同様に、蛍光シグナルの変動を正規化するためにパッシブリファレンス色素を含めることができます。
注意すべき点は、すべてのqPCRミックスに5’-3’エキソヌクレアーゼ活性を持つポリメラーゼが含まれているわけではないということです。そのため、このタイプの検出が必要な場合は、適切なプローブ検出ミックスを選択する必要があります。
ヘアピンプローブ(モレキュラービーコン)
ヘアピンプローブ(モレキュラービーコン)検出では、加水分解プローブと同様に、クエンチャー分子と蛍光分子を運ぶように共有結合的に修飾された 1 つのプローブを利用します。ただし、この配列はヘアピン構造を形成するようにも設計されており、共有結合で修飾された塩基が近接するため、プローブがヘアピンを形成してターゲットに結合していないときに、強力な蛍光色素消光が確実に行われます。このタイプのプローブには、クエンチャーと蛍光分子の相対位置が異なる様々なバリエーションが存在し、ヘアピンが変性段階で分離されるかアニーリング段階で分離されるかによってシグナル発生のタイミングが異なります1。
デュアルハイブリダイゼーションプローブ
デュアルハイブリダイゼーションプローブは、qPCRターゲットに特異的な2つのプローブの存在を前提としています。どちらもターゲットの同じ鎖上の隣接するセグメントに連続的に結合するように設計されており、最初のプローブは 3’ 末端に FRET ドナー分子を運ぶように共有結合修飾され、2 番目のプローブは 5’ 末端に FRET アクセプター分子を運ぶように共有結合的に修飾されています。両方のプローブがターゲットに結合し、ドナー分子が適切な励起波長によって刺激されると、FRET を介して励起エネルギーがアクセプター分子に移動し、次に、対応する波長で蛍光が発せられ、qPCR 機器によって記録されます。
アクセプター分子とドナー分子は、両方のプライマー上で逆の順序で設計できます。ただし、両方のプローブがターゲット分子に結合した際に、それらが互いに隣接している必要があります。デュアルプローブシステムでFRETが十分に機能するには、アクセプター分子とドナー分子が10Åの半径内に存在する必要があります。つまり、プローブはターゲット分子上で互いに隣接して(3bp未満で)結合する必要があります。
プローブベースの検出のメリット
- 色素ベースの検出と比較して、特異性が高くなる
- プライマー セットとプローブを多重化する機能
プローブがターゲット分子にアニーリングした後にのみ蛍光が発生し、非特異的産物によってシグナルが発生しないため、より高い特異性が実現されます。これは、結合する二本鎖DNA配列に関係なくシグナルを生成するインターカレーティング色素とは異なります。
この特異性と異なる蛍光色素の使用可能性により、このシステムは、単一のqPCR反応ミックスで複数のターゲットを同時に検出するマルチプレックス反応の構築に使用できます。これは、スペクトル的に異なる蛍光色素で標識された特異的オリゴヌクレオチドプローブを添加するだけで実現され、各プローブは1つのサンプル中の別々のターゲットについて特異的な定量情報を提供します。マルチプレックス化は、研究および診断の両面において、サンプル中の複数のターゲットを効率的かつ正確に定量できる非常に有用なアプローチです(図4)。
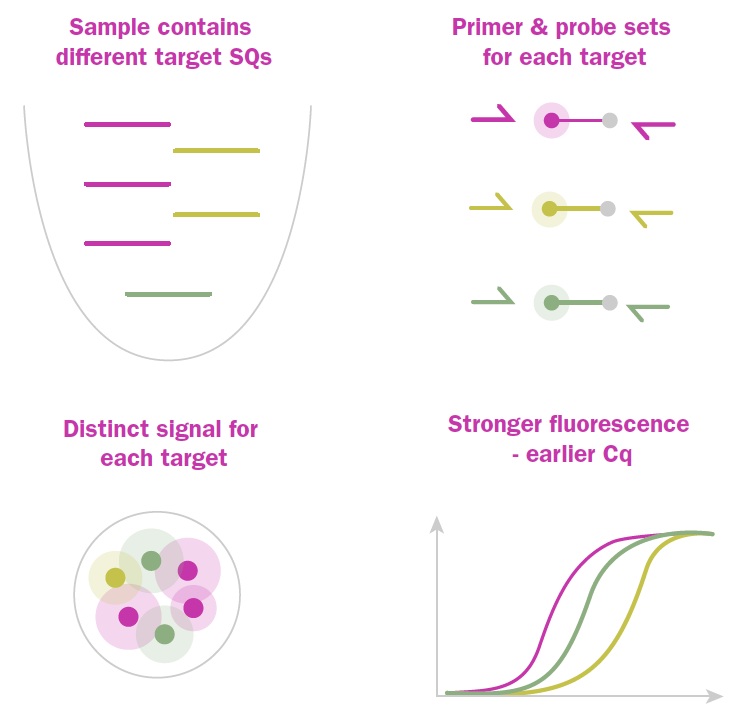
図4:異なる蛍光プローブを用いたマルチプレックスqPCRシグナル生成
プライマーダイマー、ヘアピン、またはプローブの二次構造は、プローブベースの qPCR にも影響します。これは、そのような構造の形成により、実際のターゲットに結合するために利用できるプライマーとプローブの量が減少するためです。この競合は通常、Cqの遅延を引き起こします。
色素ベースの検出方法とプローブベースの検出方法には、どちらも特有の利点と欠点があります。特に重要なのは、どちらの方法も過剰なサイクリングに起因するPCRアーティファクトの影響を受ける可能性があることです。ただし、色素ベースのqPCRは、このようなアーティファクトの影響で誤ったデータを生成する可能性がより高くなります。「トラブルシューティング」のセクションでは、これらの問題について説明します。
| 検討事項 | 色素ベースの検出 | プローブベースの検出 |
| コスト | 低コスト: 酵素ミックスとターゲットごとにプライマー1組 | 高いコスト: プローブミックス、プライマーペア1組、ターゲットごとに少なくとも1つの高純度オリゴプローブ(修飾オリゴ) |
| 実験デザイン | 簡単: ターゲットごとに配列特異的なプライマーを選択し、同様のアニーリング温度を確保 | より複雑: 適切なアニーリング温度と高い特異性を持つプライマーとプローブを設計 新しいターゲットごとに追加のプローブが必要 |
| 特異度 | プライマーダイマーや非特異的産物を含む、あらゆるdsDNAからの非特異的シグナルの可能性 | 高い特異性を有し、プローブ配列と高い配列相同性を持つ産物のみがシグナルを生成 プライマーダイマーによるCt(Cq)遅延の可能性 |
| マルチプレックス化 | 不可 | 可能 |
| 融解曲線解析 | 可能 | アニーリングプローブ(例:モレキュラービーコン)のみ可能 加水分解プローブ(例:TaqMan®)では不可 |
| 汎用性 | 容易に適応可能 新しいプライマーペアを使用すると、新しいターゲットを実験に含めることが可能 | 各ターゲットには、プライマーに加えて特定のプローブが必要 プローブとプライマーは、同様のパラメータを持つ必要あり |
| 推奨アプリケーション | 遺伝子発現解析 種多様性解析 (16S または同様のもの) HRM解析 類似のアプリケーション… | 診断用qPCR ジェノタイピング/アレル量解析 高い特異性を必要とするその他のアプリケーション |
表1:色素ベースとプローブベースの qPCR の長所と短所
3. ターゲット選択、プライマーとプローブ設計
3.1 ターゲット
ターゲット領域は、qPCR 反応の効率と特異性を最大限に高め、生物学的に意味のある明確な結果を生成するように選択する必要があります。つまり、
- ターゲット領域は、テンプレート サンプル内で固有である必要があります。言い換えれば、サンプル中の宿主生物または他の汚染生物のゲノムまたはトランスクリプトームの他の領域と相同性が低い必要があります。
- ターゲットの GC 含有量は、理想的には 30 ~ 70% である必要があります。
転写ターゲットに複数のスプライスバリアントがあることがわかっている場合、バリアント間の区別が望まれない場合を除き、異なるエクソンのスプライスジャンクションにまたがるようにターゲット領域を選択する必要があります。これにより、異なるスプライスバリアントを異なるアンプリコン長に基づいて検出することが可能になります。アンプリコン長は、qPCR後に評価することも、qPCRの前に同じプライマーペアを用いて標準的なPCRで評価することもできます。
非常に類似した転写産物または遺伝子の場合、プライマーが最も多様性の高い領域に位置し、その配列が差異を示すヌクレオチドで終結するようにターゲットを選択する必要があります。これは、類似性の高い転写産物の 5 ‘ または 3’ UTR、または類似性の高い gDNA ターゲットのイントロンに存在する可能性があります。
3.2 プライマーの設計
プライマーは、二次構造(ヘアピン、自己二量体、またはクロス二量体)を回避または最小限に抑えるように設計する必要があります。これは、インターカレーション色素ベースの qPCR 検出 (すべての dsDNA 構造がシグナルを生成し、二次構造が分析にアーティファクトを導入します) とプローブベースの qPCR 検出 (プライマーが反応において制限試薬となり、効率と感度が低下します) の両方にとって重要です。実際、反応効率を最大化するには、二次構造を回避することが重要です。最適な設計を実現するには、適切な二次構造予測ソフトウェアを常に使用する必要があります。市販の Beacon プライマー設計プログラムの無料オンライン バージョン3や Primer34 などのツールは、オンラインで入手できます。ゲノムおよびトランスクリプトームデータベースに対するPrimer BLASTは、可能な限り最適なプライマーセットを特定し、高い特異性を確保するために、複数の適切なプライマーペアを用いて実施する必要があります。
これらの一般的な考慮事項に加えて、さらに次のような設計上の特徴があります。
- プライマーの長さは18~25bpにする必要があります
- 両方のプライマーのアニーリング温度が最適になるように、プライマーの Tm 差は 2°C を超えてはなりません
- 3’ 末端に 1 つまたは 2 つの G または C 残基(3’ アンカーと呼ばれる)を持つプライマー配列を選択します。
- プライマーは、アンプリコン長が80~250bpとなるように設計する必要があります。理想的には、この範囲の短い方です。qPCR で信頼性の高いデータを得るには、PCR 効率が可能な限り 100% に近づく必要があり (ターゲットの完全な 2 倍化を示します)、増幅効率はターゲットの長さと逆相関します。
- 真核生物の遺伝子転写物 (cDNA) を検出するためのプライマーを設計する場合、エクソンのスプライス結合部分にまたがるプライマーを選択することをお勧めします。これにより、2 つのエクソン間に未切除のイントロンが含まれるため、混入したゲノム DNA が qPCR 中に増幅されることはありません。
- 新しいターゲット用のプライマーを設計するときは、2 ~ 3 組のプライマー ペアを選択してラボでテストします。
- プライマーは、qPCRミックスのメーカーが推奨する量で反応液に添加してください。各プライマーの最終濃度は通常300~900 nMですが、反応を最適化するためにこの範囲内で調整することができます。
3.3 プローブの設計
オリゴヌクレオチドプローブを設計する際の考慮事項はプライマーの場合と同じですが、いくつかの追加パラメータがあります。
- プローブ配列は可能な限り特異的である必要があり、テンプレートサンプル内のオフターゲット領域またはプライマーと交差反応しないようにする必要があります。
- 増幅中はプローブがテンプレートに結合したままでなければならないため、プローブのアニーリング温度は増幅プライマーの温度より 5 ~ 10 °C 高くする必要があります。
- グアニン残基は蛍光体を消光する可能性があるため、配列の GC 含有量は 30 ~ 80% とし、G よりも C を多く含むように設計する必要があります。
- 極めて重要なことは、プローブ設計中は 5’G 残基を避けるべきであるということです。この残基には蛍光体が加水分解後も結合したままであり、DNAポリメラーゼによるプローブ分解後にシグナルが消光する原因となります。
- プローブは高品質かつ高純度である必要があります(例:HPLC精製または同等の精製)。プローブを用いたqPCR実験を初めて行う場合は、様々なサプライヤーと精製方法を試し、アプリケーションに最適なものを特定する必要があります。
- 使用する蛍光色素とクエンチャー分子は適合性がある必要があります(表2)。選択した蛍光色素は、使用するqPCR装置で検出可能でなければなりません。FAM色素は最も適合性の高い蛍光色素であり、ほとんどのサプライヤーから入手可能です。そのため、シングルプレックスプローブに適しています。マルチプレックス反応用のプローブを設計する場合は、以下のセクション3.4「マルチプレックスqPCRの設計」のヒントを参照してください。
- 反応中の個々のプローブの最終濃度は、qPCRミックスメーカーの推奨量から始めて、100~500 nMの範囲にする必要があります。反応を最適化するために、必要に応じて濃度を調整してください。
| 蛍光色素 | 蛍光波長(nm) | クエンチャー |
| FAM | 520 | ZENTM-Iowa Black® FQ, Black Hole Quencher®-1 |
| ATTO™ 488 | 522 | |
| TET | 539 | |
| ATTO™ 532 | 554 | |
| JOE | 555 | |
| HEX | 555 | |
| TYE™ 563 | 563 | Iowa Black® FQ, Black Hole Quencher®-2 |
| Cy®3 | 564 | |
| ATTO™ 550 | 575 | |
| TAMRA | 583 | |
| ATTO™565 | 591 | |
| ROX | 608 | |
| ATTO™ Rho 101 | 609 | |
| TEX 615 | 613 | |
| Texas Red®-X | 617 | |
| TYE™ 665 | 665 | |
| Cy®5 | 668 |
表2:蛍光色素とクエンチャーの互換性
3.4 マルチプレックスqPCRの設計
マルチプレックス qPCR 反応は、プローブベースの検出にさらなる利点をもたらします。異なる蛍光色素を持つプローブを用いることで、同一反応で複数のターゲットを定量することが可能になります。
マルチプレックスqPCRのメリット
- 1 回の反応で複数のターゲットを定量できるため、実験のスループットが向上
- ノーマライザー遺伝子がターゲットと同じ反応でアッセイされる場合のピペッティングエラーの減少
- 1 回の反応からより多くの情報を抽出できるため、サンプル消費量が削減される
しかし、マルチプレックス化には、慎重な実験設定によって最小限に抑えるべき課題も存在します。シングルプレックスにおけるプライマーとプローブの設計、そしてターゲットの選択について説明した考慮事項は、マルチプレックスqPCRにも当てはまります。
マルチプレックスプライマー設計の考慮事項
- より効率的なターゲットが試薬を消費して他のプライマー/プローブセットの効率をさらに低下させないように、同様の効率を持つターゲットとプライマーを選択する必要がある
- プライマーとプローブは、異なるセット間でクロスダイマーを形成してはならない
- 各プローブに選択された蛍光色素がスペクトル的に異なるように、特別な注意を払う必要がある
個々のターゲットの存在量を評価するには、各プローブとプライマー セットの効率をシングルプレックス アッセイでテストし、次にマルチプレックスでテストする必要があります。したがって、1 つのターゲットが他のターゲットと 3 Cq 以上異なる場合、およびマルチプレックス アッセイで特定のセットの効率が低下する場合は、問題となっている反応を最適化するための措置を講じる必要があります。または、追加のプライマーまたはターゲットを検討する必要があります。
4. プライマーの検証と反応効率の計算
4. プライマーの検証と反応効率の計算
プライマー検証のキーポイント
- プライマーが特異的であることを確認する
つまり、選択したサンプルタイプで予想される固有のターゲットを増幅し、予期しないアンプリコンを生成しないことを確認する - プライマーが二量体を生成しないことを確認する
(プライマー設計中に実験的に、また in silico で検証する必要がある) - qPCR におけるプライマー反応効率を計算し、それが 90 ~ 110% であることを確認する
プライマーの検証
プライマーの検証は、実験で使用されるのと同じタイプのアンプルで実行する必要があります。例えば、ゲノムDNAを試験する場合はgDNAサンプル、cDNAサンプルを使用する場合は逆転写RNAサンプルを使用する必要があります。プライマーは、予測されるプライマーペアのTm付近のアニーリング温度勾配を用いた標準PCRまたはqPCRで試験し、増幅アーティファクトを生じずに使用できる温度範囲を評価する必要があります。
完了後、たとえテストqPCRで融解曲線解析を実施済みであっても、反応はアガロースゲル電気泳動をする必要があります。潜在的なアーティファクトとしては、プライマーダイマーやヘアピンに起因する低分子量で短い(100 bp未満)産物、およびミスプライミングによる非特異的なターゲット増幅に起因する長いアンプリコンなどが挙げられます。アーティファクトが認められる場合は、アニーリング温度範囲を可能な限り高くし、サイクル数を最大35~38サイクルに減らしてください。それでもアーティファクトが発生する場合は、サンプル調製方法(例:RNAサンプルに残留DNAが存在しない、またはサンプルのコンタミネーションが発生していない)を見直した上で、ターゲットの異なる領域に対して新しいプライマーを設計する必要があります。
プライマーをテストする場合、精製されたターゲットまたはプラスミドベクターにクローン化されたターゲットの使用は推奨されません。アーティファクトの明らかな原因は、実験サンプル中に存在する非ターゲット核酸のミスプライミングまたはオフターゲットプライミングであるためです。精製されたターゲットが検証に使用される場合、この情報は失われます。ただし、精製されたターゲットは反応のポジティブコントロールとして役立ちます。
反応効率
反応効率は、既知量のターゲットを含むサンプルの希釈系列に対して qPCR を実行し、各サンプルで観察された Cq に対してターゲット量の対数をプロットし、得られたデータ ポイントを通じて線形方程式をカーブ フィッティングすることによって計算されます。これにより、図8に示すようなデータが生成されます。
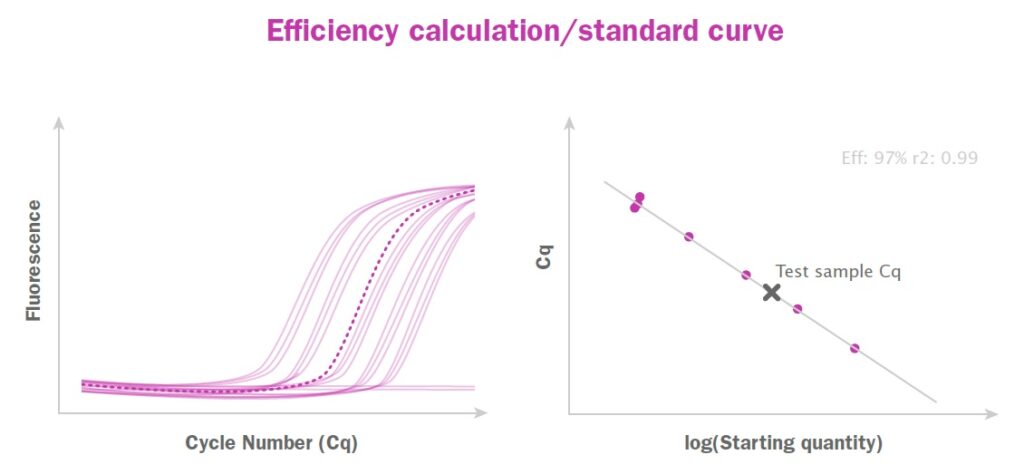
図8:標準曲線の増幅プロットとその結果の効率曲線
qPCR効率の計算
各リファレンスサンプルについて観察された Cq に対するターゲット量の対数をプロットします。
最適な直線の傾きを計算します。
Slope = ΔCq / Δlog(sample amount)
次の式を使用して、qPCR の対数増幅の線形部分におけるプライマーペアの反応効率を計算します。
Efficiency (E) = 10(-1/slope)-1
前述したように、プライマー ペアの効率は 90 ~ 110% でなければなりません。100%未満の効率は、通常の条件下では各サイクルでターゲット配列が完全に倍加することは稀であるという生化学的事実を反映しています。100%を超える効率は通常、反応阻害物質が存在する場合に観察され、各希釈度の効率を異なる程度に変化させます。
標準曲線から計算される追加パラメータは、相関係数 (R2) です。相関係数は、曲線がデータ ポイントにどの程度適合しているかを示し、理想的には完全に適合していることを示す 1 (または 100%) である必要があります。また、そのプライマー ペアを使用した qPCR の検出限界を理論的に予測する Y 切片もあります (図 8)。
5. qPCR における定量方法
名前が示すように、qPCR では既知の配列を持つ核酸ターゲットの定量化が可能です。qPCR実験の設定方法に応じて、DNAとRNAの両方のターゲットを定量化でき、絶対単位、質量単位、またはテンプレート分子のモル数(コピー数)で定量情報を得ることができます。また、qPCRは相対的な定量データも提供します。この場合、測定単位は、同一サンプル中の1つ以上の他のターゲット分子に対するターゲット分子の量、および/またはリファレンスサンプル中に存在するターゲットの存在量に対する相対的な量を反映します。
どちらのアプローチも、生物学的に意味のある情報を計算する際に Cq 値を利用します。ただし、qPCR データに関するすべての比較および統計分析は、Cq 値自体ではなく、上記の定量化方法のいずれかから導出された単位で行うことが重要です。これは、Cq 値は単なる機器の出力であり、直接比較すると誤った結論につながる可能性があるためです。さらに、Cq値は対数スケールでの差を反映しており、1つのCq差は実際のターゲット存在量の2倍の差を反映します。したがって、サンプル間の Cq の小さな差が、ターゲット量の大きな差を反映している可能性があります。
以下ではそれぞれの定量化アプローチを、それぞれの長所と短所とともに詳細に説明します。
5.1 絶対定量化
qPCR でターゲット分子の絶対定量を達成するには、既知量のターゲット分子を含む一連の標準リファレンスサンプルが必要です。標準サンプルは、過去の実験でターゲット分子の存在量が推定されている実験サンプル、または精製されたターゲット分子(PCRおよびゲル抽出によって生成されたもの、あるいはベクターにクローニングされたもの)のいずれかです。ただし、前述のように、後者のアプローチは推奨されません。
種類に関係なく、標準サンプルを使用して、生物学的に関連するターゲット存在量を検出不可能なレベルにまで広げるのに十分なデータ ポイントを含む希釈シリーズを生成します。これらのサンプルは、評価対象の実験サンプルと共にqPCR実験に使用されます。次に、標準サンプルの希釈系列を使用して、反応効率の計算について前述したように標準曲線を作成します。次に、標準曲線に最もよく適合する線形方程式を使用して、実験的に観察された Cq に基づいて実験サンプル中に存在するターゲットの量を推定します (図8)。
絶対定量化のメリットとデメリット
メリット
- このアプローチの利点は、同じ研究室内であっても異なる研究グループ間であっても、異なる実験間でデータを明確に比較できることです。また、個々の細胞内のターゲット存在量を正確に定量化できるため、研究中のシステムをより完全に把握できます。
デメリット
- 逆に、絶対定量法を用いる最大の欠点は、(i) 標準サンプル(精製またはクローン化)の作成と標準曲線の作成に必要な時間の増加、および(ii) 評価対象となるターゲットテンプレートごとにそれに応じたコストの増加です。これは、多くのターゲットが並行して評価される場合、実験のスループットが低下することも意味します。
したがって、このアプローチは、実験に1つまたは2つのターゲット分子の絶対値が必要な場合に最適です。逆に、多数のサンプルにわたって複数のターゲットを評価する場合、このアプローチの効率は大幅に低下します。
5.2 相対定量化
このアプローチでは、qPCRを用いて、ターゲット分子の存在量をリファレンス分子(複数可)に対する相対的な存在量を推定します。相対定量は主に遺伝子発現研究で用いられますが、以下に述べる基準を満たす場合は他の用途にも拡張可能です。この手法の根拠は、あらゆる生物において特定の遺伝子がほとんどの実験条件下で安定した発現レベルを維持し、そのため実験的処理を施しても転写産物の存在量に大きな変動がないという経験的観察に基づいています。これらはハウスキーピング遺伝子として知られており、その存在量が実験内のすべてのサンプルで同様であるため、リファレンス遺伝子として適しています。
相対定量を達成するために、実験のすべてのサンプルについてターゲット遺伝子とリファレンス遺伝子の両方の Cq 値が qPCR によって測定され、ターゲット遺伝子の存在量がリファレンス遺伝子の発現の割合として計算されます (図9)。
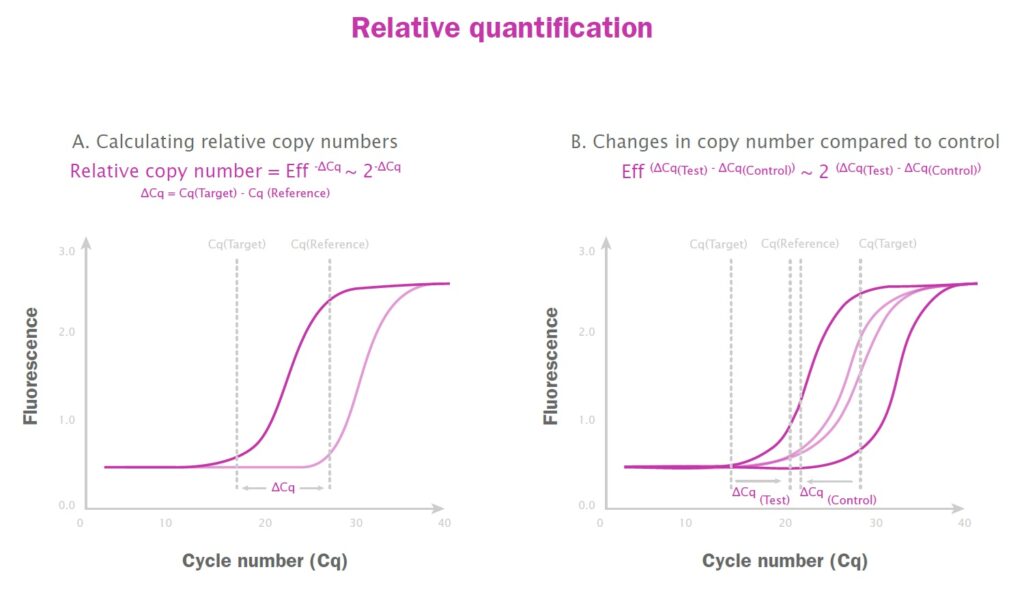
図9:1 つのサンプル (A) およびテスト対コントロール サンプル (B) のリファレンス遺伝子を使用した相対定量
この値はターゲット分子の絶対数に関する情報は提供しませんが、サンプル間で類似しているリファレンス遺伝子の値に正規化されているため、サンプル間でターゲットの存在量を比較することができます。このアプローチで有意義な実験データを取得するには、選択したリファレンス遺伝子が、特定の実験で使用されるすべての条件と処理において実際に安定していることを確認することが重要です。これは、事前の実験、トランスクリプトミクスデータセットから確立、または標準曲線を用いた絶対定量によって検証される必要があります。哺乳類で一般的なリファレンス遺伝子には、ACTB(アクチンB)、GAPDH(グリセルアルデヒドリン酸デヒドロゲナーゼ)、18S rRNA遺伝子などがあります5。哺乳類のリファレンス遺伝子のリストはオンラインで入手できます6。植物における一般的なリファレンス遺伝子は、ユビキチンリガーゼ(シロイヌナズナでは UBQ10、マメ科植物では UBQ1)、TBC(チューブリン C)、EF1a、および PP2A(タンパク質ホスファターゼ 2 A)です7。ここに記載されていない他の遺伝子は、異なる生物、組織の種類、または実験条件に適している場合があります。
サンプル間の正規化にはリファレンス遺伝子が最もよく用いられますが、相対定量実験では他の安定したパラメータも使用できます。例えば、サンプル質量、qPCRにおける総核酸量、あるいは選択された実験条件下で変化せず、実験誤差やバイアスを最小限に抑える類似のサンプル特性などが挙げられます。
相対定量化のメリットとデメリット
メリット
- qPCRで相対定量を用いる主な利点は、試薬コストや実験セットアップにかかる時間を最小限に抑えながら、複数のターゲットを並行してハイスループットで解析できることです。実験に新しいターゲットを含めるのに必要なのは、検証済みのプライマー ペアだけです。研究対象の処理条件下での発現レベルが安定していることを検証すれば、同じ参照遺伝子を用いて複数のターゲット遺伝子の発現を評価できます。一方、絶対定量では、検査対象となるすべてのターゲットについて完全な標準曲線が必要となります。
デメリット
- 相対定量の欠点としては、相対的な値であるため、異なる実験間、異なる研究グループ間、あるいは異なる論文間での発現値の比較にリスクを伴うことが挙げられます。また、同一のターゲット遺伝子に対し異なるリファレンス遺伝子を用いた相対定量実験は、直接比較できません。さらに、このアプローチは高精度ですが、検証後であってもリファレンス遺伝子の発現レベルに予期せぬ変動が生じるというバイアスが生じる可能性があります。さらに、生成されるデータは相対的であるため、遺伝子発現レベルという文脈においてのみ意味を持ち、実際のターゲット遺伝子存在量の単独の値として使用することはできません。
6. 実験のセットアップ、データ分析、MIQE
6.1 qPCR 実験のセットアップ
qPCR を最も効率的に使用して生物学的に意味のあるデータを生成するには、従うべき手順が数多くあります8。
1.どのターゲットを分析に含めるかを検討します。
2.必要な実験条件、処理、および時点を列挙します。
3.絶対定量を行う場合は、研究対象のターゲットが既知量で存在するリファレンスサンプルを用意してください。相対定量を行う場合は、研究条件下で安定な適切なリファレンス遺伝子を特定してください。リファレンス遺伝子が入手できない場合は、サンプル量に応じて、外因性のリファレンスターゲットをサンプルに添加することを検討してください。
4.ターゲット遺伝子およびリファレンス遺伝子のプライマーを設計および検証するか、文献で特性評価されているプライマーを使用します。後者の場合でも、プライマーの効率と特異性をテストし、それらが報告通りに機能することを確認する必要があります。
5.各実験条件で使用する生物学的複製 (異なる個体、細胞培養、またはコロニーの数) および技術的複製 (同じテンプレート サンプルでの反応の数) の数を計算します。必要に応じて、統計的検出力検定を使用して、ターゲット発現における統計的に有意な変化を特定するために必要な生物学的複製の最小数を確立します。生物学的複製の絶対最小値は 3 であり、これは文献で一般的に報告されていますが、これは代表的で生物学的に意味のある結果を保証するのに決して十分ではなく、盲目的に適切であると選択すべきではありません。技術的複製の数は通常 2 ~ 3 ですが、強い技術的変動の原因が疑われる場合は、それ以上になる可能性があります。絶対定量実験を行う場合は、標準曲線サンプルを含めることを忘れないでください。通常、標準サンプル希釈系列は、ターゲット分子コピー数 108 から 100 までの範囲であり、テンプレートなしコントロール (NTC) が含まれます。ただし、上限は経験的に推定し、必要に応じて変更できます。
6.必要なコントロールサンプルを特定します。反応ミックスの活性をテストするためにポジティブコントロールサンプルを含めます。プライマーペアごとにテンプレートコントロール(NTC)を含め、すべてのプレートで実行する必要があります。同じ実験で複数のプレートを実行する場合は、解析中のプレート内変動を最小限に抑えるために、プレート内コントロールとして使用できるポジティブコントロールサンプル/ターゲットを検討してください。cDNAサンプルを使用する場合は、結果を歪める可能性のあるgDNAやその他のテンプレートの混入の影響を計算するために、逆転写コントロール(NRT)を含めないでください。
7.最も正確な生物学的結果が得られるサンプルを選択してください。例えば、器官全体または生物体全体からの核酸の一括抽出は、局所レベルまたは組織レベルでの転写変化をマスクする可能性があります。特定の細胞や組織の種類を解剖または選択することで、ターゲットレベルにおける局所的な差異が明らかになる場合があります。可能であれば、サンプルは同一条件下で採取し、急速凍結して抽出まで-70℃未満で保存してください。
8.サンプルの取り扱いと調製が最適であり、可能な限り最高品質のサンプルが得られるようにしてください。RNA は特に分解しやすいため、遺伝子発現や診断分析に重大な影響を与える可能性があります。DNA はより安定していますが、サンプルが保護されるように注意する必要があります。A260/A280 > 1.8 および A260/A230 > 2.0 の高純度の核酸が得られる精製方法を選択してください。アガロースゲル電気泳動またはキャピラリーフロー分析 (例: RIN Bioanalyzer) により、精製サンプルの完全性を確認してください。RNA サンプルは、コンタミネーション DNA を除去するために DNase で処理し、エンドポイント PCR または下流の qPCR に NRT コントロールを含めることで DNA がないことを確認する必要があります。すべてのサンプルに対して同じ方法を使用して、総 DNA または RNA を定量してください。異なる方法で生成された値を使用しないでください (例: 同じ実験内で UV 吸光度と色素ベースの定量結果を比較しないでください)。
9.qPCRプレートの準備ができたら、汚染源や技術的エラーを最小限に抑えるようにしてください。可能であればクリーンベンチ内、または少なくともラボ内の専用エリアで作業してください。可能な場合はリキッド ハンドリング ロボットを使用します。各プライマー セット (および該当する場合はプローブ) のマスターミックスを準備し、すべてのサンプルに必要なマスターミックスが余っていることを確認します。
10.サンプルを qPCR マスターミックスと組み合わせる場合は、大量のサンプルを使用するのが理想的です。一般的に使用されるサンプル量は、qPCR 反応の 10~20% です。これは、20 μL の総反応量中 2 ~ 4 μL となります。可能であれば、マスターミックスとサンプル希釈液がサンプル量に対して約60~40%の割合で混合されるように調製してください。これにより、同じピペットをピペッティング量の中間値でセットアップに使用できるため、ピペッティングエラーを最小限に抑えることができます。また、マスターミックスとサンプルの混合もより良好になります。ウェルが適切に閉じられ、シールされていることを確認し、適切な遠心分離機でプレートをスピンダウンし、ウェル内に気泡がないことを確認してください。気泡があると、サーマルサイクリング中に蛍光異常が発生します。
11.製品マニュアルの推奨に従ってサーマルサイクリングプログラムを実行し、実験用プライマーの検証時に最適と判断したアニーリング温度を選択してください。必要に応じて、サーマルサイクリングプログラムの最後に融解曲線測定ステップを含めてください。融解曲線測定の正確な設定は機器によって異なりますが、通常は付属ソフトウェアにプリセットされています。該当するマニュアルをご参照ください。
12.取得後、増幅プロットや融解曲線に明らかな問題がないか、またNTCおよびNRTコントロールにアンプリコンが存在しないか、結果を評価してください。解析前に、これらの問題が発生した場合は記録しておいてください。この時点で問題が見つかった場合は、「トラブルシューティング」セクションを参照して適切な解決策を見つけてください。通常、すべてのテストサンプルの測定後、5サイクル以上経過してもNTCに生成物が蓄積している場合は許容範囲内とみなされ、解析を正常に行うことができます。ただし、このような生成物がターゲットとは区別され、テストサンプルに存在しないことを確認することをお勧めします。多くの場合、これらはプライマーダイマーです。
6.2 分析と査読品質のデータレポート
qPCR の実行に明らかな問題がない場合 (上記のポイント 11 を参照)、実行データをエクスポートして分析できます。ほとんどの qPCR 機器はしきい値を自動的に定義し、すべてのサンプルの Cq 値を生成します。このしきい値は手動で高くまたは低く調整できますが、Cq 値に誤りが生じる可能性があるため、経験の浅いユーザーには推奨されません。分析からサンプルを除外する上限 Cq 値を設定します。35~40 サイクルを超える Cq 値は、入力テンプレートの量が非常に少ない (反応開始時のターゲット コピー数が 10 未満) ことを意味します。このような濃度では、最初の qPCR サイクルでの増幅はほぼ確率的であり、技術的な変動が大幅に増加します。多くのサイクルを伴うより長いプログラムでは、より多くのアーティファクト、プライマーダイマー、ミスプライミング生成物、不完全なターゲットフラグメントも導入されるため、より高い Cq 値が必ずしも真のターゲット量を反映するとは限りません。一般に、40 を超える Cq 値には疑問がありますが、分析に含めるか除外する前に、ターゲットの特異性を慎重に評価する必要があります。絶対定量法と相対定量法のどちらを行うかによって、正確な計算方法は異なります。以下に、それぞれの方法の手順を概説します。
6.3 絶対定量データ分析
絶対定量では、各技術的複製の Cq が標準曲線と比較され、そこから入力テンプレートの量が計算されます。最新のソフトウェアのほとんどは、標準曲線を自動的に作成し、計算と対数変換を行い、初期サンプル量を適切な単位(ng、コピー数、または単位体積あたりのモル数)でエクスポートします。これらの単位は線形スケールであるため、生物学的レプリケートの記述統計量を直接計算し、選択した存在量単位を用いて直接統計解析を実行できます。
ただし、エクスポートされた Cq データの分析を自分で実行したい場合は、次の手順を実行する必要があります。
1.エクスポートされた Cq 値と各リファレンスサンプル希釈の開始量の log10 値を使用して標準曲線をプロットします。
2.データに最もよく適合する線を見つけます。Microsoft Excel、R、Pythonスクリプトなど、様々なプログラムで行うことができます。この最もよく適合する直線を表す方程式を取得し、その傾きと Y 切片に注目します。
3.各実験サンプルの技術的複製の平均値を計算します。
4.ステップ 2 で生成された標準曲線方程式を使用して、ステップ 3 の各実験サンプルの平均 Cq を入力し、各サンプルの log10 開始量を計算します。
5.ステップ 4 で計算された log10 開始量に 10 を累乗して、実際の実験サンプルの開始量を導き出します。
6.記述統計を計算し、実際の開始量に対して統計解析を実行して、実験的結論を導き出します。
6.4 相対定量データ分析
ΔCq 法
ΔCq 法では、ターゲット遺伝子とリファレンス遺伝子の Cq値 の差が式 (1) に従って計算されます。
ΔCq = Cq(Target )–Cq(Reference) (1)
そして、ターゲット遺伝子の相対発現は次のように計算されます。
E-ΔCq (2)
ここで、Cq(Target ) と Cq(Reference) は、それぞれ同一サンプル中のターゲット遺伝子とリファレンス遺伝子についてqPCRで得られた実験的Cq値であり、E は反応効率です。理想的なシナリオでは、ターゲット遺伝子とリファレンス遺伝子の両方の反応効率が100%であるため、E = 2となります。これはLivakとSchmittigenが最初に用いた手法であり、実験効率が高い場合には依然として貴重な実験情報を提供します9。しかし、反応効率が100%を大きく下回る場合には不正確であると批判されてきました。
実験的に計算された効率も式(1)に使用できますが、ターゲット遺伝子とリファレンス遺伝子の両方で同じか非常に類似している場合に限られます。実験的に計算された効率(標準曲線経由)の代替として、各反応の効率を数学的に計算する方法があります(各サンプルの増幅プロットにおけるCqデータと対数蛍光データの線形回帰分析)。このような反応効率計算用のフリーソフトウェアが利用可能であり10、qPCR機器に付属する一部の市販ソフトウェアパッケージにも含まれています。この計算された効率は、式(1)のEの値として使用できます。反応効率(理論的または実験的に計算されたもの)がターゲット遺伝子とリファレンス遺伝子の両方で同程度であると仮定すると、平均効率を式(1)のEの値として使用できます。このアプローチは、ターゲット分子とリファレンス分子の計算された効率が大きく異なる場合には使用すべきではありません。どの方法を用いるかにかかわらず、生成された相対発現値により、サンプル間でのターゲットの発現の比較だけでなく、同じリファレンス遺伝子に正規化された他のターゲットの発現との比較も可能です。したがって、これは複数のターゲットを用いた遺伝子発現解析を行うための非常に汎用性の高いツールです。
ΔΔCq法
ΔΔCq法では、リファレンス遺伝子に加えて、正規化のためのサンプルを使用します。そのために、式 (1) に従ってすべてのサンプルの ΔCq 値が計算され、その後、以下の式 (3) に従って、各テスト サンプルの ΔCq がリファレンス/ノーマライザー サンプルの ΔCq に正規化されます。
ΔΔCq = ΔCq(Test) – ΔCq(Control) (3)
コントロールサンプルと比較した遺伝子発現の倍率変化は次の式で求められます。
E-ΔΔCq (4)
ここで、ΔCq(Test) と ΔCq(Control) はそれぞれ、テストサンプルと、他のすべてのサンプルを正規化するコントロールサンプルまたはリファレンスサンプルの ΔCq 値です。E は、ターゲット遺伝子とリファレンス遺伝子の両方の計算された平均効率です。これは次のようにも表すことができます。
2-ΔΔCq (5)
リファレンス反応とターゲット反応の両方に対して完全な効率を仮定した場合。この方法では、すべてのサンプルのターゲット遺伝子の相対発現値が、選択されたコントロールサンプルと比較した倍率変化として与えられます。コントロールサンプルの倍率変化値は常に 111 になります。実験では、これは通常、未処理または未改変の遺伝子型サンプルになります。ターゲットの相対量のこの値は、実験全体のすべてのサンプルの同じターゲットの値と比較できますが、リファレンスサンプルではすべてのターゲットの値が 1 であるため、異なるターゲット間では比較できません。したがって、正規化の追加レベルにより、実験全体での単一のターゲットの変化のより堅牢な推定が可能になりますが、同じサンプル内であっても異なるターゲットの相対量について推論を行うことはできません。
相対定量を行う場合は、次の手順を実行する必要があります。
1.各生物学的サンプルのすべてのターゲットについて、反応効率とすべての技術的複製 Cq の平均を計算します。これらの平均値は、以降のすべてのステップで使用します。
2.各生物学的複製におけるすべてのリファレンス遺伝子の平均 Cq を計算し、リファレンス遺伝子 Cq として使用します。これを使用して、式(1) に従ってそのサンプル内のすべてのターゲット遺伝子の ΔCq を計算できます。
3.すべての生物学的サンプル中の各ターゲットの正規化された相対発現は、式(2)に従って計算することができます。
4.ΔCq 発現値については、所定の実験的処理について、すべての生物学的複製の正規化された発現値の平均を計算します。この値は、その処理におけるターゲット遺伝子の正規化発現レベルの平均です。
5.ΔΔCq 法を使用する場合は、各テスト サンプルについて式(1)からΔCq 値を計算します。コントロール条件におけるすべての生物学的複製の平均を計算します。
6.コントロール条件のこの平均相対発現を使用して、式(3)に従ってすべての生物学的複製の ΔΔCq 値を計算することにより、すべての試験条件における発現を正規化します。
7.式(5)を用いて、コントロールサンプルの平均値と比較した変化率を計算します。また、コントロール処理における各生物学的複製の倍率変化を計算して、個々のコントロールサンプルの発現を取得します。すべてのコントロール サンプルの発現は 1 に近い必要があります。すべての生体サンプルの発現変化倍数値を Log2 変換して、標準偏差、標準誤差、信頼区間を計算し、統計分析を実行します。
選択した定量化方法に関係なく、統計分析方法は実験の実行後ではなく、実験のセットアップ中に決定する必要があります。実験での十分な再現性を確保するために、この段階で統計的検出力テストも実行する必要があります。そうすることで、結果として得られる結論は意味があり、実際の生物学的事象を代表するものになります。統計分析を行う前に、対数変換された発現データに対して正規性テストと歪度テストを実行して、データが正規分布していることを確認する必要があります。対数変換後もデータが正規分布していない場合は、ノンパラメトリック検定を検討してください。適切な統計検定と検定要件に関する詳細は、統計学または生物学者向け統計学の教科書、またはウェブサイトを参照してください。
6.5 データレポート
実験を行う前に、査読付きジャーナルにqPCRデータを掲載するために必要な最小限の実験情報量を把握することが重要です。定量的リアルタイムPCR実験(MIQE)の掲載に必要な最小限の情報を提供するためのガイドラインが公開されており、科学界で広く受け入れられています12。必須ではありませんが、掲載するかどうかに関わらず、これらのガイドラインに従うことをお勧めします。これにより、結果の再現性が向上し、データの質が向上し、標準化によって他の研究者に実験内容を明確に伝えることができます。MIQE基準のチェックリストは、以下をご覧ください。
MIQE報告基準チェックリスト
(E:Essential・必須、D:Desirable・この情報を報告することが望ましい)
実験デザイン
- 実験群と対照群の定義 (E)
- 群あたりの個体数と技術的反復回数 (E)
- アッセイの実施者 (D)
- 著者の貢献への謝辞 (D)
サンプル
- 説明 (E)
- 処理したサンプルの容量/質量 (D)
- ミクロまたはマクロダイセクション (E)
- 処理手順 (E)
- 凍結した場合、方法と保存期間 (E)
- 固定した場合、どのような方法で、どのくらいの期間で保存したか (E)
- サンプルの保存条件と保存期間
(特にFFPE1サンプル) (E)
核酸抽出
- 手順および/または機器 (E)
- キット名および変更内容 (E)
- 使用した追加試薬の入手先 (D)
- DNaseまたはRNase処理の詳細 (E)
- コンタミネーション評価(DNAまたはRNA)(E)
- 核酸定量 (E)
- 機器および方法 (E)
- 純度(A260/A280、オプションでA260/A230)(D)
- 収量 (D)
- RNAの完全性:方法/機器 (E)
- 3’および5’転写産物のRIN/RQまたはCq (E)
- 電気泳動トレース (D)
- 阻害試験(Cq希釈、スパイク、その他)(E)
逆転写
- 完全な反応条件 (E)
- RNA量および反応容量 (E)
- プライミングオリゴヌクレオチド(遺伝子特異的プライマーを使用する場合)および濃度 (E)
- 逆転写酵素および濃度 (E)
- 温度および時間 (E)
- 試薬メーカーおよびカタログ番号 (D)
- 逆転写酵素使用時および使用しない場合のCqs (D)
- cDNAの保存条件 (D)
qPCRターゲット情報
- 遺伝子記号(E)
- 配列アクセッション番号(E)
- アンプリコンの位置 (D)
- アンプリコンの長さ (E)
- インシリコ特異性スクリーニング(BLAST等)(E)
- 偽遺伝子、レトロ偽遺伝子、または他の相同体?(D)
- 配列アライメント (D)
- アンプリコンの二次構造解析 (D)
- 各プライマーのエクソンまたはイントロンによる位置(該当する場合) (E)
- ターゲットとなるスプライスバリアントは? (E)
qPCRオリゴヌクレオチド
- プライマーシーケンス (E)
- RTPrimerDB識別番号(D)
- プローブシーケンス (D)
- 変更があった場合の場所とその内容 (E)
- オリゴヌクレオチドの製造業者 (D)
- 精製方法(D)
qPCR検証
- qPCRプロトコル
- 反応条件の詳細 (E)
- 反応容量およびcDNA/DNA量 (E)
- プライマー、(プローブ)、Mg2+、dNTP濃度 (E)
- ポリメラーゼの種類および濃度 (E)
- バッファー/キットの種類およびメーカー (E)
- バッファーの正確な化学組成 (D)
- 添加剤(SyGreen、DMSOなど) (E)
- プレート/チューブのメーカーおよびカタログ番号 (D)
- サーモサイクリングパラメータの詳細 (E)
- 反応セットアップ(手動/ロボット) (D)
- qPCR装置のメーカー (E)
qPCRプロトコル
- 最適化の証拠(グラジエントから)(D)
- 特異性(ゲル、配列、融解、消化)(E)
- SyGreenの場合、NTCのCq(E)
- 傾きとy切片を含む検量線(E)
- 傾きから計算したPCR効率(E)
- PCR効率または標準誤差の信頼区間(D)
- 検量線のR2(R2)(E)
- 直線ダイナミックレンジ(E)
- 検出限界におけるCq変動(E)
- 範囲全体のCI(信頼区間)(D)
- LOD(検出限界)(LOD)(E)
- マルチプレックスの場合、各アッセイの効率とLOD(E)
データ解析
- qPCR解析プログラム(出典、バージョン)(E)
- Cq決定方法(E)
- 外れ値の識別と処理(E)
- NTCの結果(E)
- 参照遺伝子の数と選択の妥当性(E)
- 正規化方法の説明(E)
- 生物学的反復試験の数と一致率(D)
- 技術的反復試験の数と段階(逆転写またはqPCR)(E)
- 反復性(アッセイ内変動)(E)
- 再現性(アッセイ間変動、CV)(E)
- 検出力分析(D)
- 結果の有意性に関する統計的手法(E)
- ソフトウェア(出典、バージョン)(E)
- Cqまたは生データのRDMLによる提出(D)
1FFPE:ホルマリン固定パラフィン包埋組織
7. アプリケーション
qPCR のアプリケーションには、DNA 分子やさまざまな種類の RNA の定量化が含まれます。以下に、最も一般的な手法をまとめます。
7.1 1-Step RT-qPCRと2-Step RT-qPCR
qPCR のこのバリアントは、RNA 分子の定量化に焦点を当てています。PCR は二本鎖 DNA テンプレートで機能するため、RNA 分子を定量化するには、まず DNA に変換する必要があります。これは、逆転写酵素または RTase と呼ばれる酵素を使用して実現され、RNA テンプレートから一本鎖相補 DNA (cDNA) を組み立てることができます。これは逆転写として知られており、qPCR と組み合わせると、逆転写 qPCR または RT-qPCR になります。従来、RT-qPCR は 2 つの手順で完了します。最初の手順では RNA が cDNA に変換され、2 番目の手順では準備された cDNA が qPCR でテンプレートとして使用されます。これにより、RNA サンプルを効率的に使用できます。通常、精製された RNA の一部のみが cDNA 合成に使用され、cDNA 調製物は複数の qPCR で複数の異なるターゲットを検出したり、転写クローニングやライブラリ構築などの他のアプリケーションに使用したりできます。
逆転写酵素が機能するには、DNAポリメラーゼのようなプライマーが必要です。一般的には、3種類のプライマー、あるいはそれらの組み合わせが使用されます。
オリゴ(dT)プライマー
オリゴ(dT)プライマーは、チミジン残基のみからなる短いオリゴヌクレオチド(15~20ヌクレオチド長)であり、ポリA含有RNA分子すべてに無差別に結合します。オリゴ(dT)プライマーのみを使用すると、通常、3’末端に富むcDNAが得られます。これは、真核生物のmRNAの3’末端にはポリA配列が存在し、第一鎖cDNAの合成効率はターゲットRNAの長さに比例して低下するためです。原核生物のRNAはポリA化されていないため、これらのプライマーは原核生物のcDNA合成には適していません。
ランダムヘキサマープライマー
ランダムヘキサマープライマーは、ランダムなヌクレオチド残基を含む6ヌクレオチドまたはその倍数からなるオリゴマーです。これらのプライマーは、十分な相補性であらゆるRNA分子に結合しますが、完全に相補的なプライマーに比べると効率は低くなります。
遺伝子特異的プライマー
遺伝子特異的プライマーは 1 つの特定の配列をターゲットとするため、ターゲット分子のみの効率的な cDNA 合成が可能になります。これは必然的に、cDNA がこの 1 つのターゲットの増幅にのみ適していることを意味します。
偏りのない効率的な cDNA 合成を実現するには、複数のターゲットを評価する一般的なアプリケーションではランダム ヘキサマーとオリゴ dT プライマーのミックスを使用するのが一般的ですが、高感度でユニークなターゲットの増幅が必要な場合 (診断用 qPCR の場合など) には遺伝子特異的プライマーが使用されます。
1ステップRT-qPCRでは、RNAサンプルとターゲット特異的プライマーを、逆転写酵素とDNAポリメラーゼの両方を含むqPCRミックスに加えます。したがって、ファーストストランド cDNA 合成と qPCR は、単一の組み合わせたサイクリング プログラムで実行されます。
このアプローチでは、反応ミックスにプライマーが含まれたターゲットのみがcDNAに変換され、増幅されます。これにより、サンプル処理のステップ数と実験時間は削減されますが、サンプル使用の柔軟性は犠牲になります。しかし、適切な反応効率の計算を行わずに誤って使用すると、特定のプライマーセットを用いた逆転写反応の効率がqPCRの増幅効率と異なる場合があり、ターゲット定量において実験誤差が生じやすくなります。このようなプライマー特異的なcDNA合成バイアスは、サンプル内のすべてのRNA分子を無差別にプライミングするランダムヘキサマーをミックスしたオリゴdTプライマーを使用する2ステップRT-qPCRではそれほど問題になりません。
逆に、ターゲット特異的プライマーの使用は、低濃度ターゲットの増幅に有利に働き、プロセス全体の感度を高める可能性があります。1ステップRT-qPCRは確かに大きな利点があり、研究ワークフローを大幅に向上させることができます。1ステップRT-qPCRと2ステップRT-qPCRのどちらを選択するかは、調査対象となるターゲットの数と種類、そして同じサンプルがどのような用途に使用される可能性があるかによって異なります。
7.2 遺伝子発現
RT-qPCRの主な用途の一つは、遺伝子発現解析です。この手法では、特定の生物、組織、または細胞種において、特定の遺伝子によって生成される転写分子(mRNA)の数を定量化できます。多くの場合、遺伝子発現は、1つまたは複数のハウスキーピング遺伝子をリファレンス分子として用いたRT-qPCRによる相対定量によって評価されます。次に、リファレンス分子に標準化した後、異なる実験条件間でターゲット遺伝子の発現を比較するように実験が設定されます。この正規化と相対定量は、通常、デルタCq(ΔCq)法またはデルタデルタCq(ΔΔCq)法のいずれかを用いて行われます。前述のように、これらの手法はどちらもqPCRを用いてリファレンス遺伝子とターゲット遺伝子のCq値を取得し、色素ベースおよびプローブベースの検出と互換性があります。
qPCRによる相対定量において重要な要素は、生物と実験に適したリファレンス遺伝子の選択です。複数の査読済み論文は、1つのリファレンス遺伝子への依存はリスクを伴い、遺伝子発現に関する誤った結論につながる可能性があることを示唆しています。これは、あらゆる実験条件下で一定レベルを維持する遺伝子の転写産物は存在しないためです。これには、いわゆるハウスキーピング遺伝子も含まれます。ハウスキーピング遺伝子は、転写産物を一定レベルまたはそれに近いレベルに維持するものの、様々な実験条件下では大きく変動します。この問題を回避するには、正規化に2つ以上のリファレンス遺伝子を使用することが推奨されます。これらの遺伝子は、qPCR実験で使用されるすべての条件下で安定的に発現していることが検証されている必要があります。検証は、公開データ、対象生物に固有の無料でアクセス可能なトランスクリプトームデータベース、あるいはリファレンス遺伝子の絶対定量のためのqPCR実験、あるいはリファレンス遺伝子のCqと反応における総入力RNA量との間に強い相関関係を確立することなどに基づいて行うことができます。
遺伝子発現解析にどの計算方法を選択するかに関係なく、サンプルとリファレンス遺伝子がソフトウェアに正しく入力されていれば、ほとんどの市販ソフトウェア パッケージとオンラインの多くのオープンソース パッケージでこれらの解析が自動的に実行されます。
7.3 種 / 対立遺伝子の豊富さ
qPCRは、生物のゲノムDNA中の様々なアレルの存在量を定量化するため、あるいは複数の生物を含む粗生物学的サンプル中の様々な種特異的遺伝子を定量化するためにも日常的に用いられています。後者は、同一サンプル中の種特異的遺伝子の代替としてしばしば用いられます。このような分析では、サンプルの種類や研究間で比較可能な情報を提供するため、通常は絶対定量が選択されます。ただし、サンプル質量、細胞数、または反応における総DNA入力量で正規化することにより、相対定量を用いることも可能です12。種特異的遺伝子の研究では、評価対象となる各株または種に特異的な1つ以上の遺伝子がターゲットとして選択されます。多くの場合、これらはそれぞれ原核生物および真核生物の16S rRNAまたは18S rRNAをコードする遺伝子、あるいは小rRNAサブユニット遺伝子と大rRNAサブユニット遺伝子の間に介在する内部転写配列(ITS)です。計画されている研究に応じて、異なるターゲットを選択することもできます。
ターゲットに関係なく、選択されたターゲット遺伝子の標準曲線を使用して、セクション5.1の 「絶対定量化」 で説明されているように絶対定量実験を実行します。標準曲線のターゲットサンプル希釈率は、ターゲットの生物学的に関連するレベルの範囲全体をカバーするように選択する必要があります。サンプル入力値も慎重に測定し、測定された絶対量を正規化するために使用する必要があります。これは、相対定量法とは異なり、サンプル間の取り扱いエラーや精製効率の違いを考慮できる共通の内部コントロールを見つけることがまれであるためです。可能であれば、実験条件全体で存在量が変わらないターゲットを含めることが推奨されます。これは内部増幅コントロールとして機能します。ただし、これは常に可能であるとは限りません。データが正規性検定および歪度検定に合格しない限り、統計解析は、対数変換を行わずに計算された存在量に対して直接実行する必要があります。
7.4 診断
qPCRによる絶対定量は、様々な病原体や疾患関連マーカーの同時検出・定量を可能にするため、診断において強力なツールとなります。これにより、患者の診断と治療において計り知れない情報が得られます。診断用qPCRは、通常、特異性を高めるためにプローブベースの定量法を採用しており、求める診断情報に応じてシングルプレックスまたはマルチプレックスで実施できます。診断用PCRでは、絶対量を報告するかどうかに関わらず、適切なネガティブコントロールとともに、常にリファレンスカーブを使用する必要があります。マルチプレックスqPCRは、複数の病原体を並行して同時に検査したり、同じ病原体の異なる配列をターゲットとする複数のプローブを使用できるため、検査の特異性を高めることができるため、特に診断用途に適しています。発表された報告によると、1回のマルチプレックス反応で10種類以上の病原体をスクリーニングできることが示唆されており、このqPCRアプリケーションは非常に強力です13。診断用qPCR実験を設定する際には、セクション3.4の「マルチプレックスqPCR設計」の推奨事項に従ってください。
さらに、サンプルをターゲットに対して陽性または陰性として識別するための診断に関連するしきい値を確立するように注意する必要があります。qPCRを診断ツールとして使用する場合、このしきい値は臨床的に検証される必要があります。これには、ターゲットの検出に使用するプライマーと、潜在的な病原体を正確に検出し、非病原性株の類似ターゲットを検出しないというターゲットの適合性の両方の観点から、選択したターゲットに対するqPCRの特異性を確立することが含まれます。さらに、検出限界(LOD)および定量限界(LOQ)は、関連する査読済み文献のガイドラインに従って確立する必要があります。qPCR検査の相対的または定量的な精度も推定する必要があり、方法の精度も確立する必要があります。これらの検査パラメータの設定方法の詳細については、関連する科学文献を参照してください。
7.5 ジェノタイピング
プローブベース qPCR のもう 1 つの用途は、ジェノタイピングのためのアレル(対立遺伝子)識別です。このプローブベース検出の特定の用途では、スペクトル的に異なる蛍光体を持つプローブを使用してマルチプレックス反応で特定のアレルをターゲットにすることで、異なる遺伝子型を識別できます。結果として得られる蛍光信号出力により、特定のサンプルにどのアレルが存在するかを識別することができます。このアプローチは、各アレルの量が計算されるのではなく、各プローブの信号強度が特定のしきい値を超えた場合に存在または不在として記録されるという点で、通常、定量的ではありません (ただし、同じ qPCR 実験からそのような情報も抽出できます)。したがって、結果は各アレルについて定性的で 2 値 (存在/不在) になります。1 つのサンプルの各アレルのデータを組み合わせることで、遺伝子型を確立できます (図 10)。
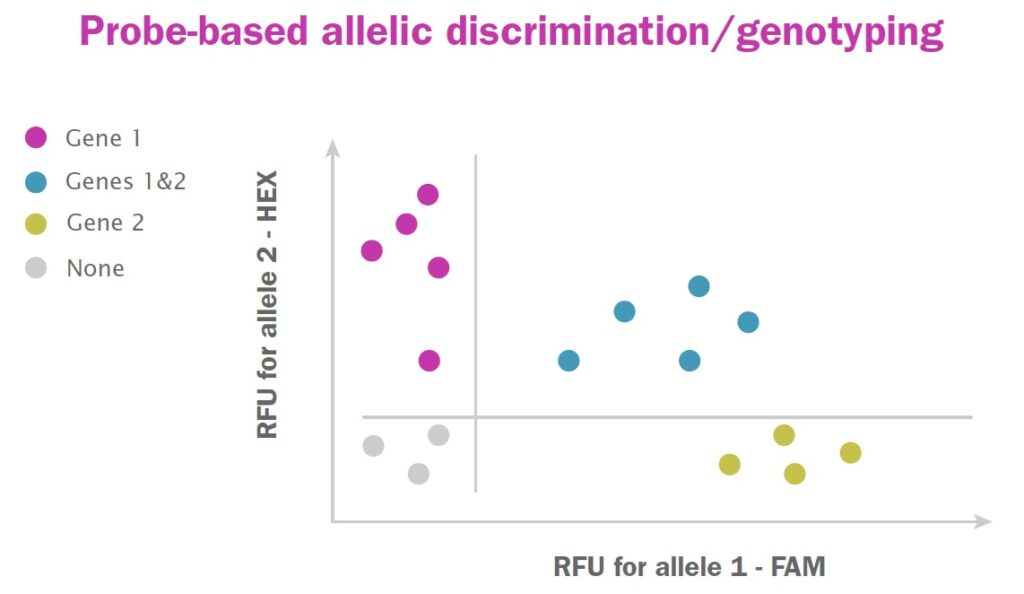
図10:プローブベースの qPCR を使用したアレル識別
たとえば、アレル A および B を持つ単一遺伝子座の単純な2アレル状態では、アレル A に特異的なプローブとアレル B に特異的なプローブの 2 つのプローブが設計され、それぞれに異なる蛍光色素がタグ付けされます。これらは、アレル A がホモ接合である個体の固有のプローブ シグナル、アレル B がホモ接合の個体の別個の固有のプローブ シグナル、およびアレル A と B の両方を保有するヘテロ接合個体からの両方のプローブからの混合シグナルを生成します。
7.6 高解像度融解曲線(HRM)分析
HRM曲線解析は、インターカレーション色素を用いたqPCRの威力を活用し、温度変化の増分をより細かく制御できる改良型サーモサイクリング装置と組み合わせることで、融解曲線解析を実行します。qPCR後の標準的な融解曲線解析では、蛍光データは0.2~0.5℃ごとに取得されますが、HRM解析では0.1℃ごとにデータを取得するというはるかに高密度なデータを取得するため、得られる融解曲線の分解能が大幅に向上します。これにより、単一の点変異(SNP)に至るまで、配列のわずかな差異でさえも、融解曲線のシフトとして記録されます。したがって、HRMは、SNP検出、アレル識別、またはターゲットバリアント検出において、プローブベースの検出法に代わる、より安価で効果的な代替手段となります。このアプローチの欠点は、融解曲線のシフトがqPCRアーティファクトの結果である可能性もあるため、特異性が低下することです(セクション2.2の「融解曲線解析の限界」で説明)。HRM解析で得られた結果は、慎重に検証する必要があります(図11)。
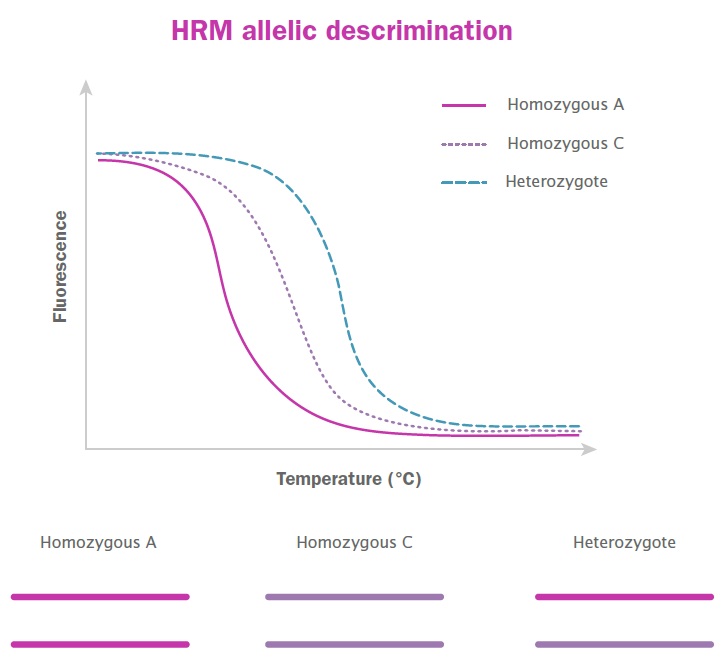
図11:SNPおよびアレル識別研究のためのHRM
8. qPCRBIO製品をテストするためのガイドライン
PCR Biosystems 社のqPCR製品または製品サンプルを初めてご使用になる際は、必ずこれらのテストを実施することをお勧めします。これらの推奨事項に従うことで、qPCRデータの信頼性が確保されます。
8.1 一般的なコメント
PCR Biosystems 社のqPCRミックスは、合計8種類のバリエーションをご用意しています。2種類の基本ミックス(レギュラーミックスとブルーミックス)があり、それぞれLo-ROX、Hi-ROX、No-ROX、またはフルオレセイン入りのミックスをご用意しています(表3上)。これらのミックスはすべて性能面で同等であり、使用するqPCR機器のパッシブリファレンス色素要件に応じて選択する必要があります。PCR Biosystems 社のプローブミックスも様々な種類のROXが含まれており、ブルー色素の有無を選択できます(表3下)。ご使用の機器に適したミックスが不明な場合は、qPCR選択ツールを使用して互換性のあるミックスを見つけるか、お問い合わせください。下記の推奨ガイドラインに関しては、これらのミックス間で区別はありません。
| qPCRBIO SyGreen Mix Blue | qPCRBIO SyGreen Mix Blue |
| Lo-ROX | Lo-ROX |
| Hi-ROX | Hi-ROX |
| No-ROX | No-ROX |
| Separate-ROX | Separate-ROX |
| Fluorescein | Fluorescein |
| qPCRBIO Probe Mix | qPCRBIO Probe Blue MIx |
| Lo-ROX | Lo-ROX |
| Hi-ROX | Hi-ROX |
| No-ROX | No-ROX |
| Separate-ROX | Separate-ROX |
表3:qPCRBIO ミックスのパッシブ色素オプション
8.2 テスト実験のセットアップ
実験の準備や、PCR Biosystems 社のミックスと競合他社製品との比較を行う際は、反応の準備に使用するプライマー、プローブ、テンプレート、希釈液、その他の添加剤はすべて、同一バッチ、できれば同一ボトルのものを使用してください。すべての反応は同じ日に、できれば同じ人が準備する必要があります。すべての比較には、同一の装置、および可能であればリキッドハンドリングロボットを使用してください。
絶対感度をミックス間で比較する場合は、ゲノム DNA を使用する必要があります。ただし、テンプレートの希釈率が低いと確率的効果が優先されるため、結果を拡大解釈しないように注意する必要があります。このようなシナリオへの対処方法については、セクション9「トラブルシューティング」で説明します。
テスト実験は次のようにセットアップする必要があります。
- テストサンプルには、4 ~ 5 段階のテンプレート希釈度とテンプレートなしのコントロール (NTC) を含める必要があります。
- 逆転写反応を qPCR とは独立して実行する場合、つまり 1 ステップ qPCR 製品を使用しない場合は、真のサンプルと、通常のサンプルから逆転写酵素を除いたものと同じ試薬をすべて含む逆転写なしコントロールも含める必要があります。
- また、テスト サンプルとコントロール サンプルごとに 3 ~ 4 回の技術的複製を使用する必要があります。
- テンプレート希釈度は、広範囲をカバーし、すべてが標準曲線に寄与できるように選択する必要があります (標準曲線は少なくとも 4 つのポイントで構成されている必要があります)。
- 各反応液量は使用する支持体の量に比例する必要があり(例:96ウェルプレートの場合は少なくとも20μL)、プレート/チューブはセットアップ中ずっと4℃に保たれます。マスターミックスも同様に保管するのが理想的です(ただし、すべてのポリメラーゼはホットスタートであるため、室温でセットアップすることも可能です)。
以下の表は、当社製品をテストする際に使用すべき推奨テスト サンプルとコントロール サンプルをまとめたものです。
| サンプル | 内容 | レプリケート |
| 希釈 1 | 例:希釈されていない参照サンプル100 | x3-4 |
| 希釈 2 | 例:希釈1の10-2希釈 | x3-4 |
| 希釈 3 | 例:希釈1の10-4希釈 | x3-4 |
| 希釈 4 | 例:希釈1の10-8希釈 | x3-4 |
| テンプレートコントロールなし (NTC) | 滅菌水をサンプルとして使用 | 希釈×3~4 1 |
| *逆転写酵素コントロール(NRT)なし | すべての試薬とサンプルが含まれますが、逆転写酵素は含まれません。 | x3-4 |
*qPCR とは別に cDNA 合成ステップを実行する場合に必要です。したがって、qPCRBIO 1-Step mixesには適用されません。
表4:qPCR 製品テストに推奨される実験セットアップ
あらゆる種類のコンタミネーションを避けるよう注意が必要です。特に、高濃度のターゲットテンプレートを含むサンプルは、NTC反応を汚染することが多いため、注意が必要です。プレートのセットアップには、クリーンベンチまたは専用のqPCRスペースを使用してください。PCRプレートは適切に密封し、各ウェルを完全に密封し、マイクロプレート遠心分離機でスピンダウンして気泡をすべて除去してください。
8.3 サイクリング
比較を実行する前に、各メーカーのガイドラインに従ってサイクリング条件を最適化し、すべてのミックスが特定のテンプレートとプライマーのセットに対して最高の性能を発揮できるようにする必要があります。
最適化されたサイクリング条件がミックス間で異なる場合は、各ミックスをその最適な条件だけでなく、他の競合他社の条件でも実行することをお勧めします。
別々に実行する場合は、同じサイクル数を使用する必要があります。
各実行の後には、同一の溶融解析を行う必要があります。
プレートが適切に密閉されていること、および反応ミックスの蒸発がないことを確認するために、各実行後にプレートを検査する必要があります。
8.4 データ分析
偽のシグナル、または反応特異性の低下を示すすべてのウェルは分析から除外する必要があります。
異なるサプライヤーのミックス間で蛍光強度レベルに差があっても、それが同程度であれば、十分な根拠がない限り、それほど重要ではありません。qPCRミックスに固有のダイナミックレンジは、このような差を説明するのに十分すぎるほどです。
異なるミックスを直接比較すると、Cq しきい値推定方法に問題が生じます。異なるミックスの蛍光レベルが異なるため、しきい値法では蛍光レベルの高いミックスの Cq 値が過小評価される可能性があります。これは、シグナルの正規化(基本的にすべてのプラトーを一致させる)によって軽減できます。生のシグナルと出力を比較して、ソフトウェアがデフォルトでこのような正規化を実行するかどうかを確認してください。ソフトウェアがカーブフィッティングから Cq パラメーターを抽出して Cq を推定できる場合、またはデータをエクスポートしてカスタムメイドの分析を実行できる場合は、蛍光レベルの違いによって生じる Cq 値間の誤った差異が修正されます。それ以外の場合は、LinRegPCR などのソフトウェアを使用して視覚化し、可能な場合は Cq 閾値を移動することで Cq 値を割り当てることができます10。
各希釈系列について標準曲線を計算する必要があります。曲線の傾き(効率)と切片から、PCR増幅の品質に関する情報が得られます。効率が2(または100%)が理想的で、これは各サイクル間でDNA量が2倍になることを意味します。これより高い効率は、セットアップのエラーまたはサンプル内の阻害物質の存在を示している可能性があります。効率が低い場合は、反応条件が最適ではないことを示唆しています。たとえば、バッファーが間違っている、プライマー設計が間違っている、dNTP、Mg2+、酵素の量が間違っている、またはいずれかの成分が分解されているなどです。切片はアッセイの感度の尺度となります。値が低いほど、感度が高くなります。ただし、これは相対的な尺度であり、同じ反応セットアップ(同じアンプリコン、同じサイクリング条件、同じ機器)を比較する場合にのみ使用する必要があります。反応が異なる効率で実行される可能性があるため、異なるサプライヤーのミックスで生成されたCq値を単一のテンプレート希釈度で比較することはお勧めしません。
融解曲線を解析し、プライマーダイマーの存在を確認する必要があります。PCR産物よりも低い温度で融解ピークが見られる場合、プライマーダイマーが形成されていることを示しています。これは特に低温希釈で顕著であり、ホットスタート活性が低いことを示しています。複数のピークは、プライマー設計の不備、または効率の悪いホットスタート手法による非特異的プライミングに起因する可能性があります。PCR産物よりも高いピークは、ゲノムコンタミネーションを示唆している可能性があります。ピークの絶対的な位置は、バッファー組成の違いにより、ミックスメーカー間で異なる場合が多く、懸念事項ではありません。
9. トラブルシューティング
qPCRでよく発生する問題は、以下に記載されています。個々の実験条件や明らかな取り扱いミス(例:試薬の添加忘れ、サンプルおよび試薬の不適切な取り扱い・保管)に起因するエラーの原因は、以下には記載されていません。
一般的に、適切な実験設計、プライマーおよびプローブのバリデーション、慎重なサンプル調製、適切なポジティブコントロールおよびネガティブコントロールの使用は、問題を予防し、エラーの原因を特定するのに役立ちます。機器の性能とは関係のない問題が継続的に発生する場合は、試薬サプライヤーにお問い合わせください。PCR Biosystems 社の試薬についてはお問い合わせください。
9.1 色素ベースのqPCRに関する問題
9.2 プローブベースのqPCRに関する問題
9.3 逆転写に関する問題
参考文献と免責事項
参考文献
1.Storts, D. R. Alternative probe-based detection systems in quantitative PCR. J. Mol. Diagnostics 16, 612–614 (2014).
2.Downey, N. Explaining multiple peaks in qPCR melt curve analysis. https://eu.idtdna.com/pages/education/decoded/article/interpreting-melt-curves-an-indicator-not-a-diagnosis (2014).
3.Premier Biosoft. Beacon Designer Free Edtion. http://www.premierbiosoft.com/qOligo/Oligo.jsp?PID=1 (2021).
4.Koressaar, T. et al. Primer3. https://bioinfo.ut.ee/primer3/ (2021).
5.Freitas, F. C. P. et al. Evaluation of reference genes for gene expression analysis by real-time quantitative PCR (qPCR) in three stingless bee species (Hymenoptera: Apidae: Meliponini). Sci. Rep. 9, 17692 (2019).
6.Pampel, J. Genomics online: Housekeeping genes. https://www.genomics-online.com/resources/16/5049/housekeeping-genes/ (2021).
7.Jin, Y., Liu, F., Huang, W., Sun, Q. & Huang, X. Identification of reliable reference genes for qRT-PCR in the ephemeral plant Arabidopsis pumila based on full-length transcriptome data. Sci. Rep. 9, 8408 (2019).
8.Taylor, S. C. et al. The Ultimate qPCR Experiment: Producing Publication Quality, Reproducible Data the First Time. Trends Biotechnol. 37, 761–774 (2019).
9.Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001).
10.Ramakers, C., Ruijter, J. M., Lekanne Deprez, R. H. & Moorman, A. F. M. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci. Lett. 339, 62–66 (2003).
11.Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, 0 (2001).
12.Bustin S.A., Benes V., Garson J.A., Hellemans J., Huggett J., Kubista M, Mueller R., Nolan T., Pfaffl M. W., Shipley G.L., Vandesompele J., Wittwer C.T., The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments, Clinical Chemistry, 55, 4, 611–622 (2009).
13.Pretorius, M. A. et al. Respiratory Viral Coinfections Identified by a 10-Plex Real-Time Reverse-Transcription Polymerase Chain Reaction Assay in Patients Hospitalized With Severe Acute Respiratory Illness—South Africa, 2009–2010. J. Infect. Dis. 206, S159–S165 (2012).
免責事項
この原稿は査読を受けていません。
このガイドは、PCR Biosystems 社およびPCR Biosystems 社製品の教育資料として作成されました。qPCRの初心者または経験豊富な研究者や実験担当者向けの入門ガイドとして作成されています。すべてのプロトコル、ヒント、推奨事項は、参照文献および著者の個人的な経験に基づいています。ただし、このガイドに従うユーザーは、自身の判断で実験を計画および実施し、自己責任で実施することを理解した上で、所属機関の健康および安全に関する手順に従ってください。PCR Biosystems 社および著者は、このマニュアルに従って、または推奨されている試薬や材料を使用して得られた結果の品質、妥当性、および解釈について責任を負いません。
ATTO™、TYE™、Texas Red®-X、Cy®、ZENTM-Iowa Black® FQ、Iowa Black® FQ、およびBlack Hole Quencher®は登録商標であり、それぞれの所有者の財産です。これらの用語は、情報提供のみを目的として本文書に記載されています。PCR Biosystems 社は、本文書においてこれらの商標の所有権を主張または暗示するものではありません。